


Abstract
The reactivity of polyphenols is due to the position of the hydroxyl groups on their aromatic nuclei. Ortho-hydroxyl groups promote oxidation while meta-hydroxyl groups induce electrophilic aromatic substitution. Both hydroxylation patterns are encountered in flavonoid structures, on the B and A rings, respectively. In addition to oxidation and electrophilic aromatic substitution, flavonoids undergo nucleophilic addition on the central C ring when it is positively charged. Reactions of the A and C rings are pH-dependent. The A ring of flavonoids undergoes a polycondensation reaction mediated by an aldehyde. The products are anthocyanin and flavanol polymers and copolymers constituted of both. Flavanol polymers are not stable and rearrange into vinyl flavanols and xanthylium pigments. Vinyl flavanols can react with the positively charged C ring of anthocyanins, yielding pyranoanthocyanins, which can also be formed from components that have a reactive double bond, such as carbonyl and ethylene bonds. The positively charged C ring primarily undergoes direct reactions. Since the positive charge on the C ring of anthocyanins and flavanols is pH-dependent, their dehydration and interflavan bond cleavage reactions are also pH-dependent. This leads to flavanol–anthocyanin (F-A+) adducts at lower pH values and anthocyanin–flavanol (A+-F) adducts above pH 3.8. Temperature seems to favor formation of the latter.
Polyphenol and Flavonoid Reactions
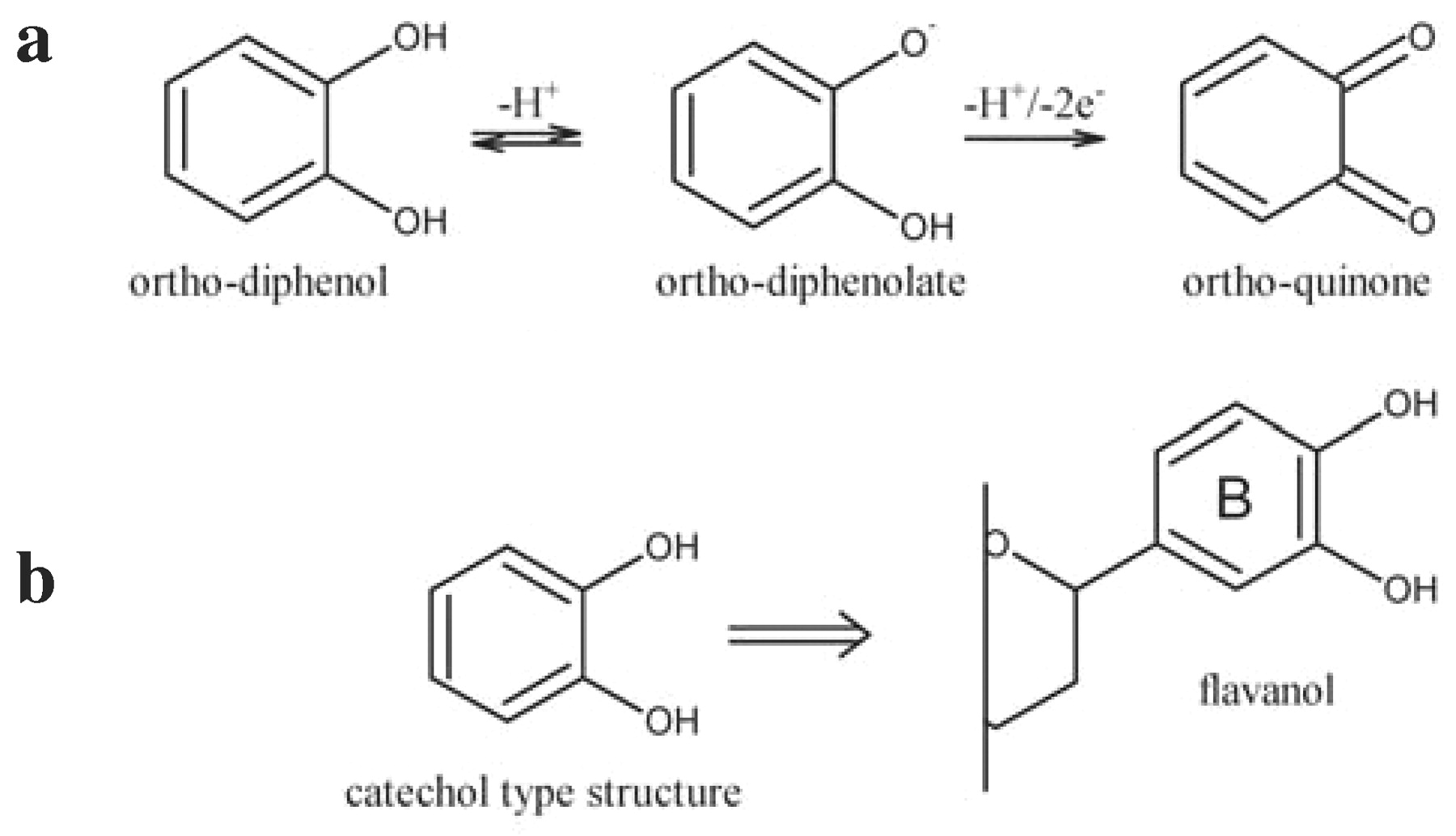
Polyphenolic structures are based on aromatic nuclei and most have more than one hydroxyl group. The presence of hydroxyl groups in the aromatic system alters its reactivity. The high electron density of the aromatic ring, enhanced by the mesomeric effect of the electron-donating hydroxyl groups, increases the electron density at particular sites of the aromatic ring (Figure 1a⇓); at the ortho-position on the carbons adjacent to the one bearing the hydroxyl group and at the para-position on the carbon opposite the carbon bearing the hydroxyl group. These activated carbons with partial negative charges are nucleophilic sites and promote electrophilic aromatic substitutions (Figure 1b⇓): the hydrogen atom attached to the partly negative carbon is replaced by an electron-deficient species (electrophile). The strength of activation depends on the number and relative positions of the hydroxyl groups. The strongest effect is achieved with three hydroxyl groups equally distributed on the phenyl ring, a resorcinol-type structure found in the A ring of the flavonoid skeleton (Figure 1c⇓).

Activated sites of phenols (a); electrophilic aromatic substitution (b); flavonoid ring (c).
The partial migration of electron density from the oxygen atom of the hydroxyl groups to the aromatic ring increases the acidity of the hydroxyl groups, facilitating the loss of a proton (H+) (Figure 2a⇓). The resulting phenolate anion oxidizes readily. When two hydroxyl groups are on adjacent carbons of the aromatic ring, the phenolic system can readily oxidize to an ortho-quinone. This hydroxylation pattern is found in catechol and the B ring of the flavanol series of flavonoids (Figure 2b⇓).

Oxidation of ortho-diphenols (a); B ring of flavonoid (b).
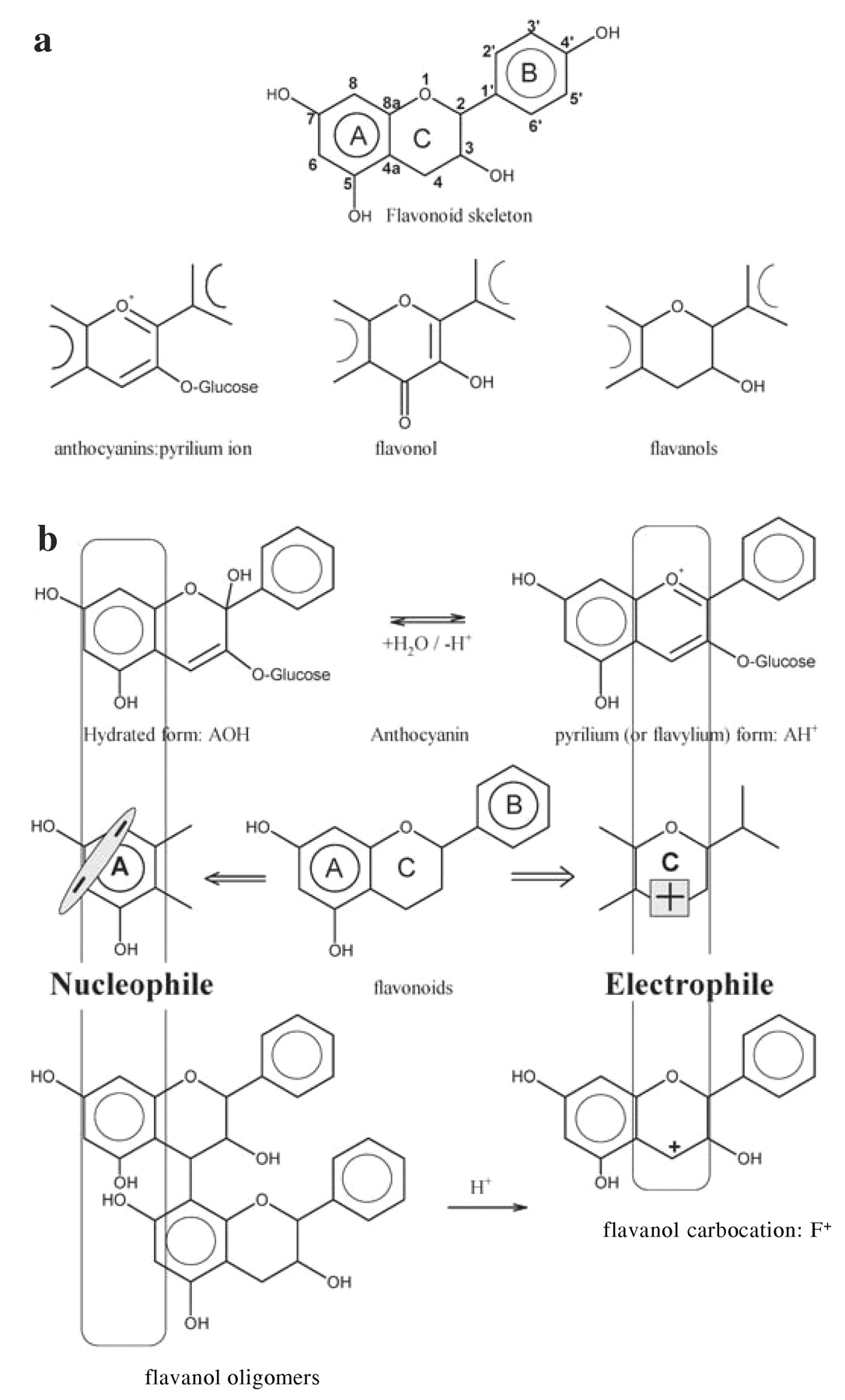
Flavonoids are the most important family among grape polyphenols, both in quantity and quality. Their structures are comprised of the two polyhydroxylated aromatic A and B rings connected through a central pyran ring, referred to as the C ring (Figure 3a⇓). The C ring can be more or less saturated; the number of double bonds (unsaturation) determines the class of flavonoids (Figure 3b⇓). The fully unsaturated C ring corresponds to a pyrilium cation and is the structure of anthocyanins, the red pigments of grapes. A fully saturated C ring gives flavanols a class that includes both monomeric and polymeric structures. The latter are usually called condensed tannins or proanthocyanidins. Cleaving the interflavan linkage (IFL) between the flavanol units of the polymeric chain under even mildly acidic conditions gives rise to highly reactive carbocations. These can further oxidize to anthocyanidins (hence the name proanthocyanidin), or they can be trapped by electron-rich chemicals known as nucleophiles. Anthocyanins are also positively charged in their flavylium forms (pyrilium structures). This positive charge on the C ring, found in anthocyanins or readily generated in proanthocyanidins, makes the C ring react as an electron-deficient species, or electrophile. In anthocyanins, this positive charge can be lost by hydration, and in proanthocyanidins it can be gained by acid-catalyzed cleavage; both processes are pH-dependent. Thus, anthocyanins and proanthocyanidins can react either as nucleophiles or electrophiles depending on pH (Figure 3c⇓). Our goal is to review various reactions of anthocyanins and flavanols.

Flavonoid skeleton and variation of flavonoid structure in C ring (a). Reactions leading to electrophilic and nucleophilic species among anthocyanins and flavanols (b).
Anthocyanin Reactions
Anthocyanins are usually represented as their red-colored forms, the flavylium cations. Because of the positive charge, flavylium cations are not very stable, and there are two ways they can yield neutral species, acid-base equilibrium and hydration.
Acid-base equilibrium.
The first mechanism is a proton transfer corresponding to an acid-base equilibrium. In this equilibrium, the cation (AH+) is the acidic molecule and the neutral quinoidal base (A) is the basic species:

This equilibrium is instantaneous (Brouillard et al. 1977a, b). The position of the equilibrium is described by the acidity constant (pKa = − log Ka) characteristic of the particular anthocyanin molecule. Thus, the relative proportions of cation (AH+) and base (A−) depend on pH. The pKa value of malvidin 3-glucoside, a major grape anthocyanin, is 4.2 (Brouillard et al. 1978).
Hydration.
The second mechanism by which flavylium cations are converted into neutrally charged species is hydration. This reaction is a nucleophilic addition of water to the pyrilium nucleus, most often at C2 and less often at C4. The products of hydration are colorless hemiketals (AOH) that are in turn partly converted into tautomeric chalcones:

The equilibrium position is described by a hydration constant (pKh = − log Kh) characteristic of the anthocyanin molecule. The hydration reaction is much slower than acid-base conversion and requires several hours to reach equilibrium. The proportion of flavylium and hemiketal forms is determined by the hydration constant to pH ratio. For malvidin 3-glucoside, estimated pKh is 2.6 (Brouillard et al. 1977a,b). At pH >2.6, malvidin-3-glucoside is mainly in the colorless hemiketal form. At wine pH (3.6), 90% of the anthocyanins are hydrated flavylium cations. In this form, anthocyanins react as nucleophiles through their A ring, like flavanols. Therefore, some color-stabilizing reactions may proceed in wine. A well-known process occurring in the plant is driven by the aromatic properties of the colored forms of anthocyanins. The planar shape and hydrophobicity conferred by the aromatic rings favor vertical stacking of the colored forms of anthocyanins between them (self-association), or with other planar structures (copigmentation), or with an aromatic residue connected to the pigment (intramolecular copigmentation) (Brouillard et al. 1989, Goto et al. 1991, Dangles et al. 1992a,b). The color properties of anthocyanins are covered elsewhere (Cheynier et al. 2006).
A second way that anthocyanins preserve their color is the conversion of anthocyanins into more stable chromophores through chemical reactions, which occurs when the cellular integrity of the grape berries is broken and the different grape constituents are mixed together. Before reviewing these reactions, it is important to examine bisulfite addition, another major reaction responsible for anthocyanin discoloration in wines.
Bilsulfite addition.
Bisulfite is used in food processing and winemaking to protect against microbial and fungal growth and to inhibit chemical oxidation. However, it makes a colorless adduct with the red forms of anthocyanins (Jurd 1963):

The sulfite dissociation constant (pKs) is 5, so the anthocyanin-sulfite adduct is stable at wine pH (3.2 to 4). Therefore, anthocyanins are trapped as noncolored bisulfite adducts in proportion to the amount of sulfite added in musts. The structure of the adducts was recently confirmed by Berke et al. (1998).
Formation of pyranoanthocyanins.
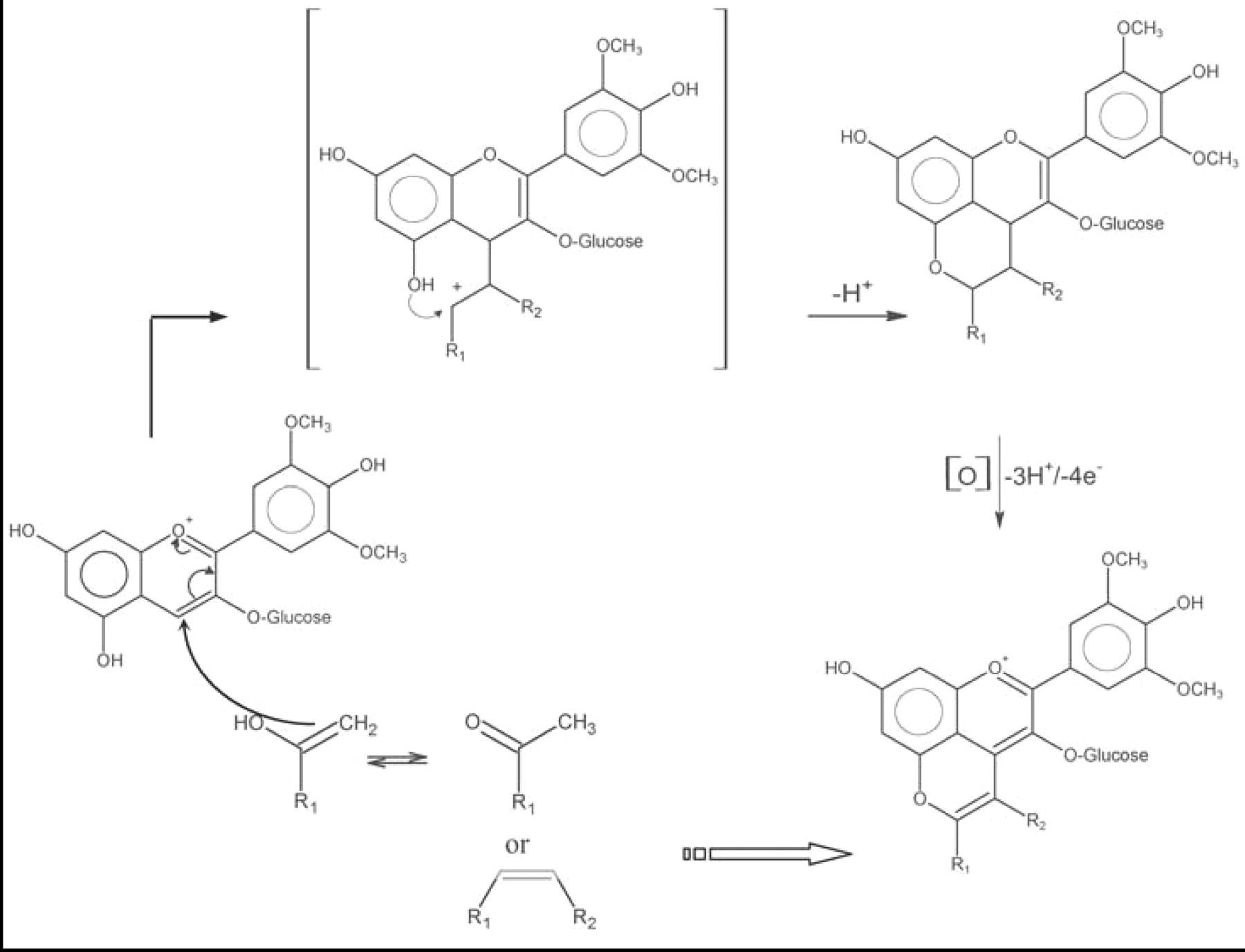
Some wine constituents and yeast metabolites react with the electrophilic portion of the flavylium cation to form new, stable pigments. An additional pyran ring forms between the hydroxyl attached to C5 and the C4 of the original anthocyanin, giving rise to a pyranoanthocyanin (Figure 4⇓). The nucleophile for this reaction must have a polarizable double bond, activated by an electron-donating group. For example, the grape p-hydroxy-substituted cinnamic acids react with the grape anthocyanins (Schwarz et al. 2003a, Schwarz and Winterhalter 2003), yielding the hydroxy-phenyl pyranoanthocyanin series (also called vinylphenol or vinylcatechol adducts; the latter also called pinotin A), which was initially obtained from the corresponding vinyl derivatives (Fulcrand et al. 1996, Sarni-Manchado et al. 1996). Another class of reactants have an enolizable double bond. Acetaldehyde, pyruvate, and other yeast metabolites formed pyranoanthocyanin-type pigments (Fulcrand et al. 1998, Benabdeljalil et al. 2000, Mateus et al. 2001), more commonly referred to as vitisin A and vitisin B (Bakker et al. 1997, Revilla et al. 1999, Romero et al. 2000a, Schwarz et al. 2003b). The particular pyranoanthocyanins formed depend on grape cultivar (Pozo-Bayon et al. 2004, Schwarz et al. 2004) and winemaking conditions such as yeast strain (Romero et al. 2000b, Morata et al. 2003). More complex structures, such as flavanol-yl pyranoanthocyanins have been detected in wines (Francia-Aricha et al. 1997, Mateus et al. 2002a,b). Their formation requires a flavanol, an anthocyanin, and acetaldehyde. Two reaction pathways have been proposed (Atanasova 2003). Flavanyl pyranoanthocyanins could result from the reaction of flavanol with a pyranoanthocyanin previously formed from acetaldehyde and anthocyanin. Alternatively, a vinyl-flavanol might react with anthocyanin. Vinyl-flavanols result from cleaving of the dimer formed by acetaldehyde-mediated condensation of an anthocyanin and a flavanol. A blue pyranoanthocyanin-like pigment with a larger chromophore has recently been identified (Mateus et al. 2003). The high stability of pyranoanthocyanins over a wide pH range has sparked growing interest in the enological study of wine color.
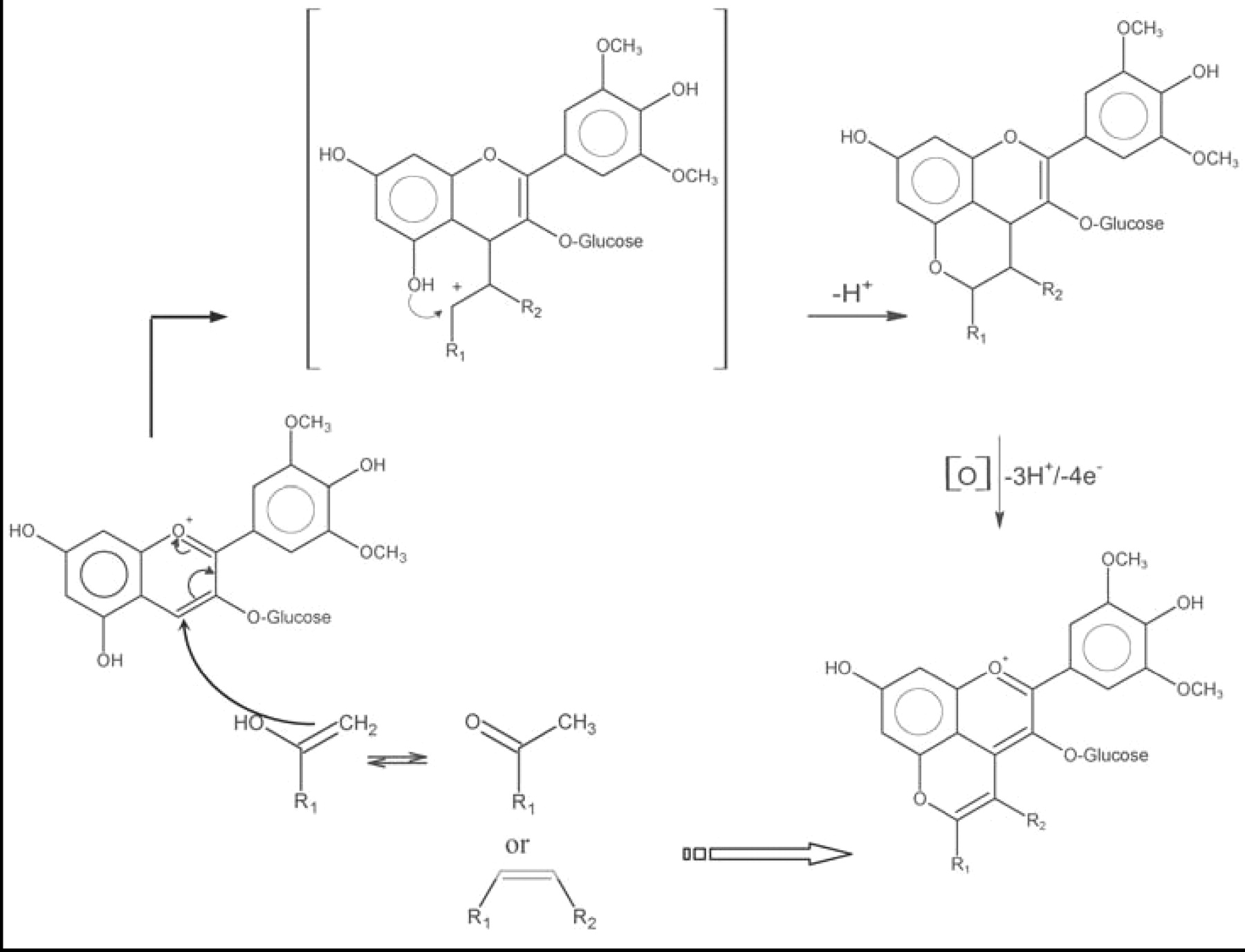
Pyranoanthocyanin formation. Yeast metabolites: acetaldehyde (R1: H), pyruvate (R1: COOH). Wine constituents: vinyl phenol (R2: H), caffeic acid (R2: COOH), flavanol (R2: H).
Polymerization reactions of anthocyanins.
Direct reaction and acetaldehyde-mediated condensation have recently been proposed as possible mechanisms of anthocyanin polymerization, yielding polyethyl-anthocyanins (A+-Et-AOH) and A-type anthocyanin polymers A-(O)-A+ (Atanasova et al. 2002a, Vidal et al. 2004). No formal NMR structural identification has yet been published for the direct anthocyanin product. However, evidence of such a reaction is available from liquid chromatography-mass spectrometry (LC-MS) analyses of a wine fraction before and after thiolysis (Salas et al. 2005). Anthocyanin polymerization is highly relevant to wine composition. How polymers form and which factors promote their formation need clarification, as do their color, colloidal properties, and contribution to wine sensory attributes.
Flavanol Reactions
Oxidation.
Oxidation of polyphenols can proceed either by transfer of one electron via the formation of a phenoxyl radical (semiquinone) intermediate or by transfer of two electrons via the formation of a quinone intermediate. Semiquinones of polyphenols are unstable and decay via dimerization or disproportionation. Disproportionation is especially common if the semiquinone contains a catechol nucleus and leads to the formation of the corresponding ortho-quinone and the regeneration of the parent catechol (Dangles et al. 2000). The ortho-quinone itself is also unstable because of its high electrophilic character and can react further in three ways. First, ortho-quinones can form dimers or larger polymers when they react with the nucleophilic parent catechol molecule. Structural identification of several oxidation dimers of catechin (Guyot et al. 1995, 1996) showed the presence of specific linkages in their structures located between the B ring of the upper unit and A ring of the lower unit of the biphenyl ether (Cφ-O-Cφ) or biphenyl (Cφ-Cφ) linkages. The biphenyl ether linkage requires a radical mechanism. Both dimer types were formed during oxidation at pH 3, indicating that semiquinone and quinone formations were in competition. At pH 6, only biphenyl linkages were identified, suggesting that the radical mechanism was no longer competitive. Second, ortho-quinones can undergo nucleophilic addition with other good nucleophiles such as amino acid residues of peptides or proteins, the cystein of gluthatione, or other phenols. Third, ortho-quinones can be oxidized by reducing species such as ascorbate or other phenols.
Flavanol oxidation in wine uses molecular di-oxygen (O2) and is thus referred to as autoxidation. The direct reaction between polyphenols and O2 is not chemically favored except under very basic conditions not found in wine. In acidic solutions, metal ions such as Fe(III) are efficient catalysts of oxidation (Hynes et al. 2001, Mochizuki et al. 2002). The resulting chain reaction of autoxidation is summarized by the following equation:

The decay of hydrogen peroxide (HO•) by traces of metal ions (a Fenton reaction) produces the very reactive hydroxyl radical (HO°) as follows:

The regeneration of Fe(III) in the Fenton reaction allows the cycle to continue.
Alternatively, hydrogen peroxide can oxidize ethanol to acetaldehyde (Wildenradt et al. 1974) in the presence of Fe(II) (Inui et al. 2004):

Although acetaldehyde is a yeast metabolite, it can be regarded as an oxidation marker (Atanasova et al. 2002b). Similarly, tartaric acid is oxidized in the presence of iron and molecular oxygen to give glyoxylic acid, which also contains an aldehyde moiety. Consequently, oxidation generates two species that react with polyphenols: direct oxidation products, such as semiquinones and quinones, and aldehydes, such as acetaldehyde.
Aldehyde polycondensation.
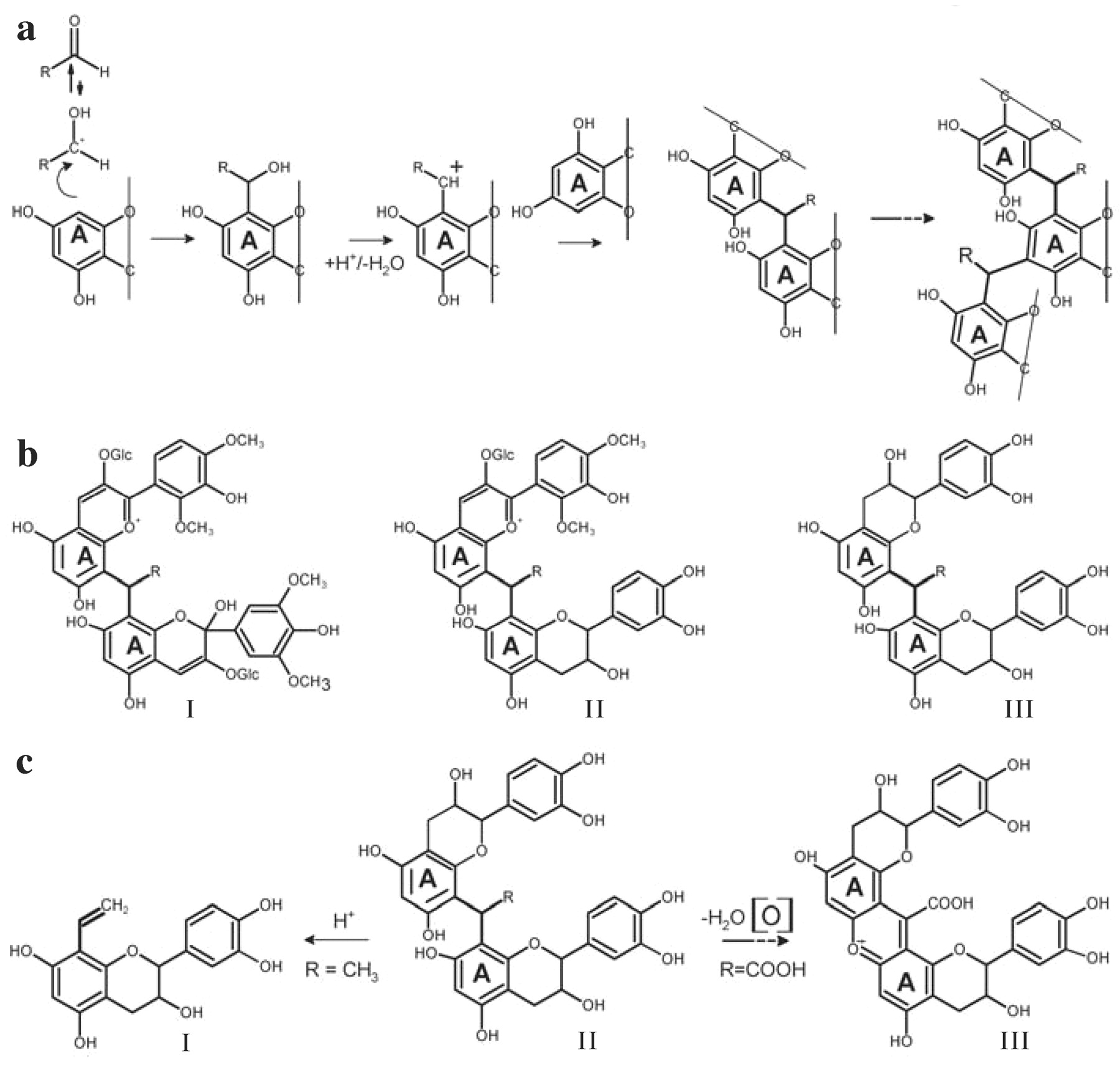
Aldehydes undergo polycondensation reactions with flavonoids. Acetaldehyde, glyoxylic acid, furfural, and hydroxymethyl furfural, which are sugar dehydration products formed during oak toasting and extracted during barrel aging, react with both flavanols and hydrated forms of anthocyanins (Es-Safi et al. 2004). The mechanism of this reaction is a Friedel–Crafts-type reaction (Figure 5a⇓). Acidic conditions allow traces of the protonated aldehyde, which is a powerful electrophile. The resorcinol-type A ring of the flavonoid structure undergoes an electrophilic aromatic substitution, where the C8/C6 hydrogen is replaced by the addition product of the protonated aldehyde, namely the corresponding alcohol. This intermediate dehydrates to give a new carbocation, which in turns reacts with a second nucleophilic flavonoid. The reaction starts again from the newly formed dimers, leading ultimately to polymers (Figure 5b⇓). When anthocyanins are involved in the reaction, polymeric purple pigments are generated. The polymers produced by flavanol aldehyde polycondensation are unstable and undergo other reactions. With acetaldehyde, the ethyl linkages generated in the polycondensated flavanol polymer are very labile and cleave into vinyl-flavanol oligomers (Figure 5c⇓). These vinyl-flavanol oligomers can then react with anthocyanins to produce the flavanyl pyranoanthocyanins.

Mechanism of aldehyde-mediated polycondensation (a). Products arising from polycondensation reactions (b): R-methine anthocyanin polymer (I); R-methine anthocyanin-flavanol copolymer (II); R-methine flavanol polymer (III). Rearrangement of R-methine flavanol polymer (c) (II) into xanthylium ions (III) and (a) vinyl-flavanols (I).
Another rearrangement can occur with polycondensated flavanol polymers, first demonstrated with glyoxylic acid (Es-Safi et al. 1999a). A condensation between two A rings of the polymer leads to a xanthylium structure (Figure 5c⇑). Xanthylium pigments have not been detected when acetaldehyde is reacted with flavanols, but the orange coloration of the initial solution is characteristic of the xanthylium chromophore. Furthermore, purple pigments were obtained along with xanthylium cations from a reaction between catechin and glyoxylic acid (Es-Safi et al. 1999b). The structures postulated for such pigments were based on the quinonoidal skeleton.
The end products of flavanol oxidation by other flavanols or by aldehydes are much more stable than the parent flavanols. Biphenyl or biphenyl ether linkages in the direct oxidation products of flavanols are particularly resistant to acid-catalyzed cleavage. The aldehyde-mediated polycondensation polymers are not as stable but can rearrange into more stable pigments, namely pyranoanthocyanins and xanthylium. The extent of oxidation and subsequent polymerization depends on several factors, including oxygen availability and the anthocyanin to flavanol ratio. Other reactions may compete with oxidation, including direct reactions between anthocyanins and flavanols.
Interflavan linkage formation and cleavage.
Inter-flavan bond formation and cleavage determine the size distribution of tannins in wines (Haslam 1980). Consequently, the size distribution of the tannin fraction is partly dependent on the ratio of flavanol monomers to polymers. As demonstrated in model wine (Vidal et al. 2002), an excess of monomer decreased the mean degree of polymerization (mDP) of the tannin fraction, while that of the control solution was unchanged. This bond-breaking and bond-making process occurs at low pH (2.0 to 3.2) (Vidal et al. 2002), but cannot occur above pH 3.8 (Salas et al. 2003). Further experiments at intermediate pH values would clarify the impact of this process on the overall size distribution of wine tannins.
Reactions between anthocyanins and flavanols.
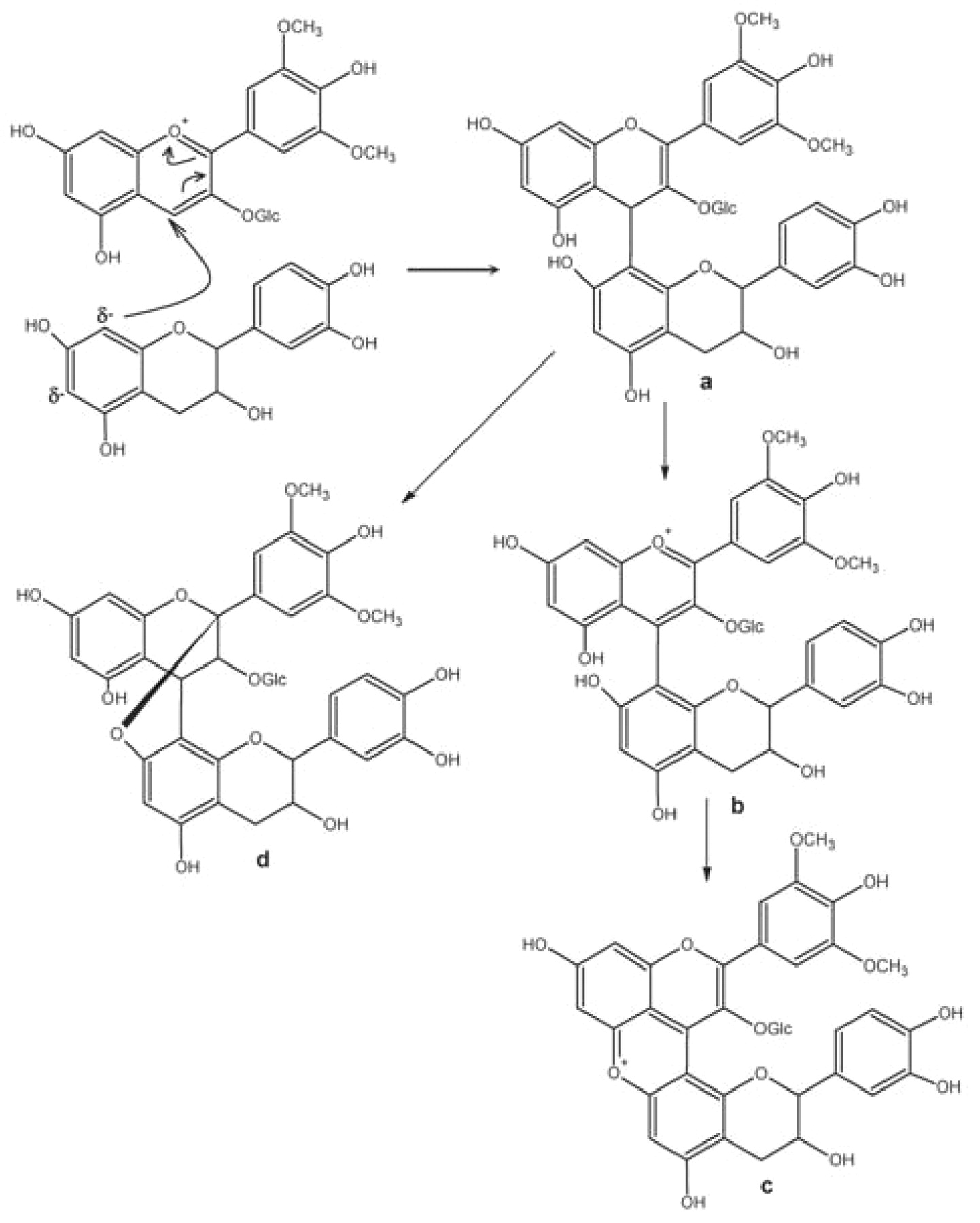
Anthocyanins in hydrated form are nucleophiles and thus compete with flavanol molecules in the bond-making process of tannin reorganization (Salas et al. 2003). Two mechanisms have been postulated for direct reactions between anthocyanins and flavanols, leading to flavanol-anthocyanin (F-A+) and anthocyanin-flavanol (A+-F) adducts, respectively. To form A+-F, anthocyanin in its flavylium form reacts as an electrophile with a nucleophilic flavanol through C8/C6, the partially negatively charged carbons of the A ring (Figure 6⇓). The first addition step leads to a colorless flavene intermediate (Figure 6a⇓), which can be oxidized into the red flavylium form (Figure 6b⇓) and then dehydrated to yield the xanthylium cation (Figure 6c⇓) (Jurd 1969, Somers 1971, Liao et al. 1992). Alternatively, the transitory flavene can evolve to a colorless cyclic adduct linked with an A-type bond [A-(4-8, 2-O-7)-T] (Remy-Tanneau et al. 2003).

Formation of the (A+-F) dimer: flavene (a); flavylium cation (b); xanthylium cation (c); (A+-F) dimer (d).
To form F-A+, the nucleophile and electrophile are reversed. The hydrated hemiketal form (AOH) of anthocyanin reacts as a nucleophile with a flavanol carbocation generated by interflavan cleavage of a tannin molecule. This produces a colorless dimer (F-AOH), which may partially dehydrate to the corresponding red flavylium (F-A+), depending on the wine pH and its own pKh.
Both reactions are pH-dependent, since the production of A+ and F+ cations requires acidic conditions. Thus, the reaction between B2 3′-O-gallate (Ec-EcG) and the major grape anthocyanin malvidin 3-glucoside (Mv3glc) was investigated at pH 2.0 in order to ensure formation of both cations (A+ and F+) and determine which mechanism is favored. The reaction was also studied at pH 3.8 to evaluate what happens at wine pH. Reaction products were obtained at both pH values, but their structures were different (Figure 7⇓). Flavanol polymers (Figure 7a⇓) and flavanol-anthocyanin (F-A+) polymers (Figure 7b⇓) were produced in the most acidic solution, showing that dimer cleavage occurred and that the low concentration of AOH (20%) did not limit formation of F-A+. At pH 3.8, A+-F type oligomers (Figure 7c⇓) were identified but flavanol polymers could not be detected, suggesting that the interflavan linkage was not cleaved at this pH.

Effect of pH on flavanol-anthocyanin reactions: flavanol polymers (a); F-A+ polymers (b); A+-F oligomers (c).
Similar experiments were conducted with a flavanol monomer instead of the flavanol dimer (Duenas et al. 2006). Under these conditions, the formation of F-A+ polymers is not possible and the A+-F dimer should be found instead. However, A+-F product could not be detected at pH 2.0 or 3.8. Instead, xanthylium chromophores were identified by their characteristic λmax (438 nm). Their masses were 633 and 453 dalton, indicating that the anthocyanin and/or flavanol units were not intact in the xanthylium pigments. Similar xanthylium chromophores were obtained from a synthetic flavylium (Escribano-Bailon et al. 1996). The authors also synthesized A+-F dimer from a homologous synthetic flavylium with no hydroxyl at the C5 position. Hence, the presence of a hydroxyl group at C5 decreased the fraction of the positive charge at C4, and thus decreased the electrophilic character of the flavylium ion.
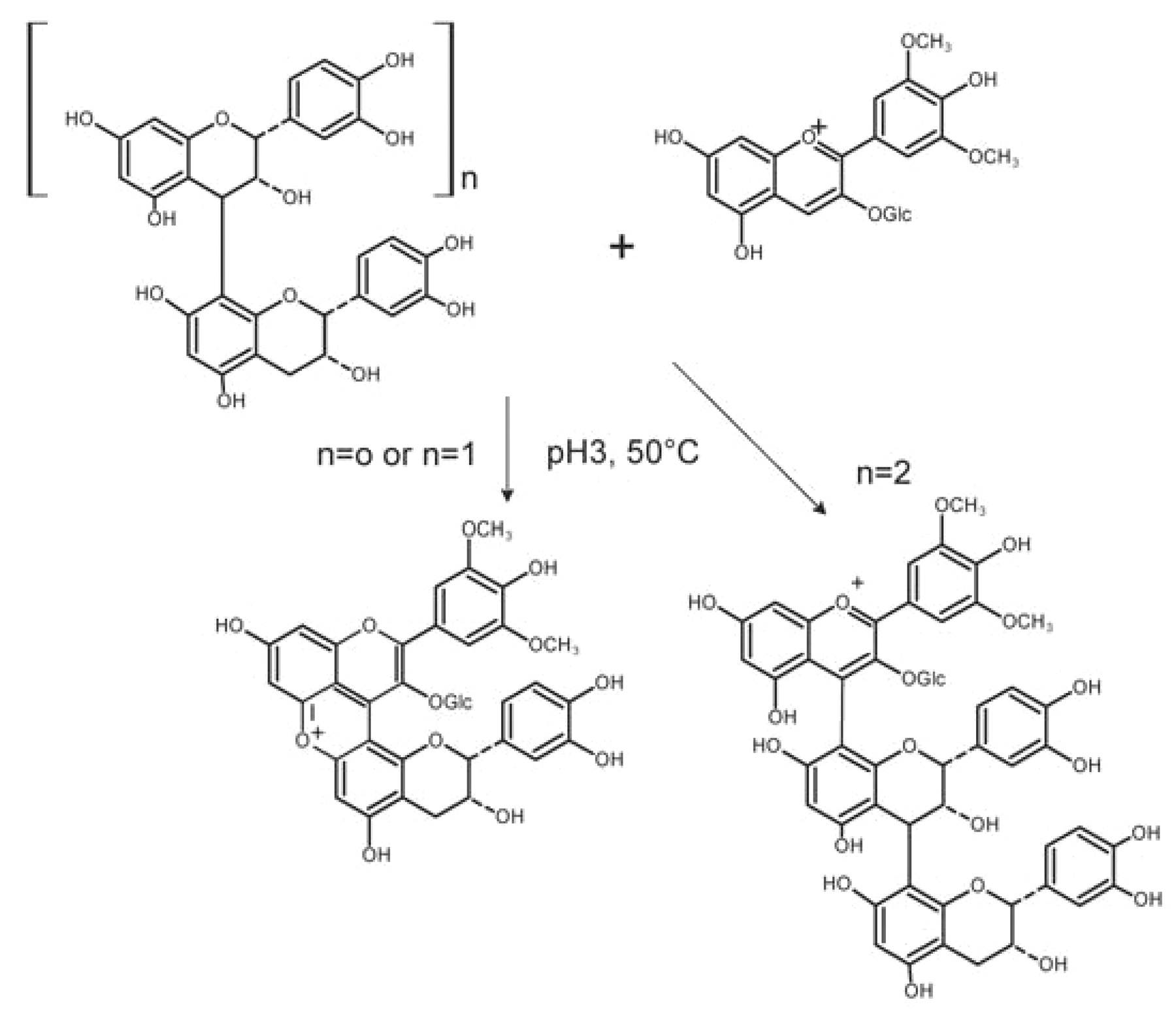
Conversely, when flavanol oligomers (di-, tri-, and tetramers) reacted with an anthocyanin at pH 3 and high temperature, only A+-F pigments were identified (Malien-Aubert et al. 2002). The absence of F-A+ polymer under these conditions suggests that the conversion of the chalcone (which is in equilibrium with the hydrated form of anthocyanin) into thermo-degradation products is faster than the reaction between hydrated anthocyanins and flavanol carbocations. In addition, A+ -F dimers generated from flavanol monomers and dimers by cleavage of the interflavan bond underwent further dehydration to give xanthylium pigments (Figure 8⇓), whereas the A+-F pigment yielded by the flavanol trimer was stable. The next step of xanthylium ion formation, dehydration, was probably blocked by the flavylium chromophore.

Effect of flavanol chain length (n) on the type of pigment arising from the A+-F reaction.
Studies emphasize the effects of pH and temperature on flavanol-anthocyanin reactions and the role of flavanol size (degree of polymerization) in determining xanthylium or flavylium products. Interflavan bond cleavage is related to the size of tannin molecule, but it depends more strongly on the structure of the tannin molecule and the nature of its constitutive units. Consequently, both types of oligomers, A+-F and F-A+, form at wine pH. The hydration constant pKh of 2.5 for the cat-Mv3glc dimer indicates color properties similar to the parent anthocyanin (Salas et al. 2004). However, larger oligomers may have different color properties because of folding, which prevents hydration of the flavylium nucleus.
Research on flavanol reactions is limited by a lack of information on the molecular weight distribution of the grape flavanol fraction and its reaction products. Thus, quantification of the various flavanol products and their relative proportions has not yet been achieved. Better methods of determining polymeric structures must be developed in order for knowledge of wine polymers to advance.
Conclusion
Numerous reactions of flavonoids occur in the course of winemaking and aging, producing a huge variety of colorless products and pigments. Their relative quantity depends on many factors (Fulcrand et al. 2004). The pH determines the percentage of the flavylium cations compared with the hydrated forms of anthocyanins. It also controls some other reaction pathways: semiquinone and quinone formation, subsequent steps in their oxidation, acetaldehyde-induced polymerization, and interflavan and ethyl bond cleavages. Temperature and flavanol structure also affect the products of direct reactions. At pH <3.8, F-A+ products were produced, but the reverse structures, A+-F pigments, were identified at high temperature (50°C). In addition, the direct reaction carried out with flavanol monomers led to xanthylium pigments instead of A+-F pigments, which were obtained with larger flavanols (dimers and trimers).
The relative proportions of anthocyanins and flavanols also affect the nature and size of wine pigments and tannins. An excess of anthocyanins significantly increases anthocyanin polymers, which are more stable than their precursors. High tannin concentration may favor direct reactions between anthocyanins and flavanols, competing with the continuous bond-breaking and bond-making of tannins themselves. Both processes change the mean degree of polymerization of the flavanol fraction. Oxygen favors autoxidation reactions. Direct reactions between di-oxygen and organic molecules such as polyphenols is not chemically possible, but they are catalyzed efficiently by traces of metal ions, especially iron. Two common oxidation reactions are oxidative flavanol polymerization and aldehyehyde-mediated polycondensations of anthocyanins and flavanols.
The pH, anthocyanin to tannin ratio, and oxygen availability vary with grapegrowing and winemaking conditions. The relative proportions of anthocyanins and flavanols in grapes depend primarily on the variety, but can be modified by environmental and cultural conditions. Winemaking practices also help determine the extraction of phenolic compounds and their subsequent reactions in wine. Maceration conditions greatly affect the relative proportion of anthocyanins and flavanols diffusing into the wine. Wine pH depends on grape maturity and fermentation, but it can be altered by addition of tartaric acid or electrodialysis. Oxygen levels in wine depend mostly on handling during winemaking. Any operation involving air contact may introduce dissolved oxygen: wine transfer, stirring, racking, stabilization, or bottling. Apart from these accidental inputs, oxygen can be introduced in a controlled manner using microoxygenation to improve the sensory character of the wine.
The use of new winemaking technologies is governed by the experience and empiricism of winemakers. Despite recent progress in wine phenolic chemistry, the exact relationship between red wine quality and grape and wine composition is still unclear. Information is needed on how structural changes such as linkage type and incorporation of anthocyanin in the tannin core affect tannin properties. Tannin properties essential for wine quality include not only aggregation and colloidal stability but also interactions with other wine macromolecules involved in astrigency such as proteins and polysaccharides. In our goal to control the quality of wine through selection of appropriate winemaking processes, a key challenge is how molecular changes in the tannin fraction affect the sensory quality of wines.
Footnotes
This article was originally presented at the ASEV 56th Annual Meeting Phenolics Symposium, 20–21 June 2005, Seattle, WA. All phenolics symposium articles were peer reviewed by two fellow presenters, and James Harbertson, Mark Downey, and Sara Spayd served as technical editors of the articles.
From the ASEV 2005 Phenolics Symposium
- Copyright © 2006 by the American Society for Enology and Viticulture
Literature Cited
Vol 57 Issue 3








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
More from this TOC section
Similar Articles
