


Abstract
The involvement of iron (Fe) in the reaction of wine with oxygen was deduced at least 80 years ago. It was observed that when wine was exposed to air, some O2 reacted rapidly, in part to oxidize the Fe to the ferric state and in part to generate what were thought to be unstable peroxides. It was proposed that it was these intermediate oxidants that oxidized wine polyphenols. In this present work the oxidation of (+)-catechin was reexamined in model wine to determine if different phases of oxidation could be observed. In the absence of sulfite, after an initial uptake of O2 very little further reaction occurred. This initial O2 uptake increased with increasing concentrations of Fe(II) and Cu(II), indicating that Fe was the initial reactant and furthermore was absent when Fe was added as Fe(III). In contrast, in the presence of sulfite, the oxidation of (+)-catechin was markedly accelerated and, again, an initial more rapid phase of oxidation, presumably due to the metals, was discernable. With (+)-catechin, insufficient O2 to oxidize all the Fe was initially taken up and formation of other oxidants such as peroxides was unlikely. Further evidence is presented that nucleophiles such as sulfite accelerate oxidation by reacting with quinones. The rate of oxidation in the presence of sulfite was found to increase in the order (+)-catechin< caffeic acid< (−)-epicatechin < gallic. It is proposed that the rate of oxidation is determined by the relative reactivity of their respective quinones. In the absence of sulfite, these additional phenols were also oxidized extremely slowly. The initial faster phase of oxidation was also observed in a white wine.
The importance of iron (Fe) and copper (Cu) as catalysts of wine oxidation was deduced at least 80 years ago. In his doctoral thesis, Jean Ribéreau-Gayon (1931) reported that Fe exists in the ferrous state (Fe(II)) in wine that had been long protected from air. However, when white wine was saturated with air, Ribéreau-Gayon found that after a few hours the concentration of dissolved O2 (initially ~6 mL/L), when measured by analysis of the extracted gases, was 0.5 to 2.0 mL/L lower than when the concentration was measured by a method based on the ability of O2 to oxidize sodium hydrosulfite to bisulfite, using indigo carmine as a redox indicator. This “missing” O2 had oxidized Fe(II) to the ferric state (Fe(III)), which Ribéreau-Gayon proposed acted as a stronger oxidant than O2 itself. He concluded that the concentration of Fe(III) depended on its relative rate of formation by the reaction of Fe(II) with O2 and its rate of reduction by wine constituents. Since Fe returned to the ferrous state as O2 concentration declined, he proposed that wine oxidation involved the redox cycling of iron acting as an intermediate oxidant. In support of this proposal, Ribéreau-Gayon showed that the rate of oxidation of white wines could be slowed and eventually stopped by the progressive removal of Fe and Cu with potassium ferrocyanide. This latter observation was recently confirmed (Danilewicz and Wallbridge 2010), although without knowledge of the earlier work at the time.
Ribéreau-Gayon (1931) calculated that the initial rapid O2 uptake by white wine was in excess of that required just to oxidize Fe(II) and proposed that some, generally between 0.2 and 1.4 mL/L, also reacted directly with wine constituents to produce unstable peroxides that were also thought to be strong intermediate oxidants. However, Émile Peynaud later noted that O2 cannot combine directly with the reducing substances of wine and can only do so with the assistance of iron salts, traces of Cu enhancing considerably its catalytic action (Peynaud 1984). That conclusion is consistent with the electronic configuration of O2 in its triplet ground state. Since O2 contains unpaired electrons, it accepts electrons singly by interacting with transition metals and free radicals and not by reacting with substances such as polyphenols and sulfite, which contain all paired electrons (Danilewicz 2003).
Ribéreau-Gayon (1931) also showed that sulfite oxidation was markedly accelerated by Fe in model wine and that its action was further enhanced with the addition of Cu. Both he and Peynaud concluded that sulfite protected wine from oxidation by reacting with O2, which remained the view for many years (see Ribéreau-Gayon et al. 2000). However, it has been argued that the rate of sulfite oxidation is far too slow to be a factor in wine oxidation and that its function is to remove the hydrogen peroxide that is produced when polyphenols are oxidized (Boulton et al. 1996).
More recent mechanistic studies of the oxidation of polyphenols in the presence of sulfite strongly support the proposal that Fe is an essential catalyst, its action markedly augmented by Cu (Scheme 1). For example, 4-methylcatechol (4-MeC) is not oxidized at a significant rate in model wine containing sulfite until Fe is added. Addition of Cu then produces a marked synergistic effect (Danilewicz 2007). Since addition of Cu(II) also markedly accelerates the rate of Fe(II) oxidation, it was proposed that Cu facilitates Fe(III)/Fe(II) redox cycling. The importance of the metals has also been demonstrated in wine, as their removal with potassium ferrocyanide slows oxidation and in white wine can prevent it entirely (Danilewicz and Wallbridge 2010). The rate of sulfite reaction depends on 4-MeC and (+)-catechin concentration in model wine and also differs for different catechols, which supports the proposal that sulfite reacts with the hydrogen peroxide that is produced when polyphenols are oxidized, not with O2 directly (Danilewicz 2007, Danilewicz and Wallbridge 2010). However, the O2:SO2 molar reaction ratio was found to be 1:2, and so sulfite reacts not only with hydrogen peroxide but also with the quinone. In the case of 4-MeC, it was shown by HPLC that ~40% added sulfite to produce the sulfonic acid and ~50% was reduced back to the catechol (Danilewicz et al. 2008). With (+)-catechin, the quinone is almost entirely reduced back to the catechol. No indication of a sulfonic acid adduct was apparent by HPLC. The net result is that O2 and sulfite are consumed but (+)-catechin concentration does not appear to change (Danilewicz and Wallbridge 2010). Consequently, it could be concluded mistakenly that (+)-catechin is not involved and that O2 and sulfite simply react together. However, sulfite oxidation is a free radical chain reaction, which is inhibited by radical scavengers such as polyphenols (Danilewicz et al. 2008). Sulfite reacts rapidly with hydrogen peroxide as in the aeration-oxidation method for its determination. It also reacts rapidly with quinones, as observed in the cyclic voltammetry of polyphenols in model systems and in wine (Makhotkina and Kilmartin 2009). Further proof that quinones are indeed formed was obtained by autoxidizing catechols in the presence of benzenesulfinic acid (BSA) in model wine. With 4-MeC the sulfone adduct was formed in 94% yield by HPLC. For (+)-catechin the sulfone adduct was extracted out and characterized (Danilewicz et al. 2008, Danilewicz and Wallbridge 2010).
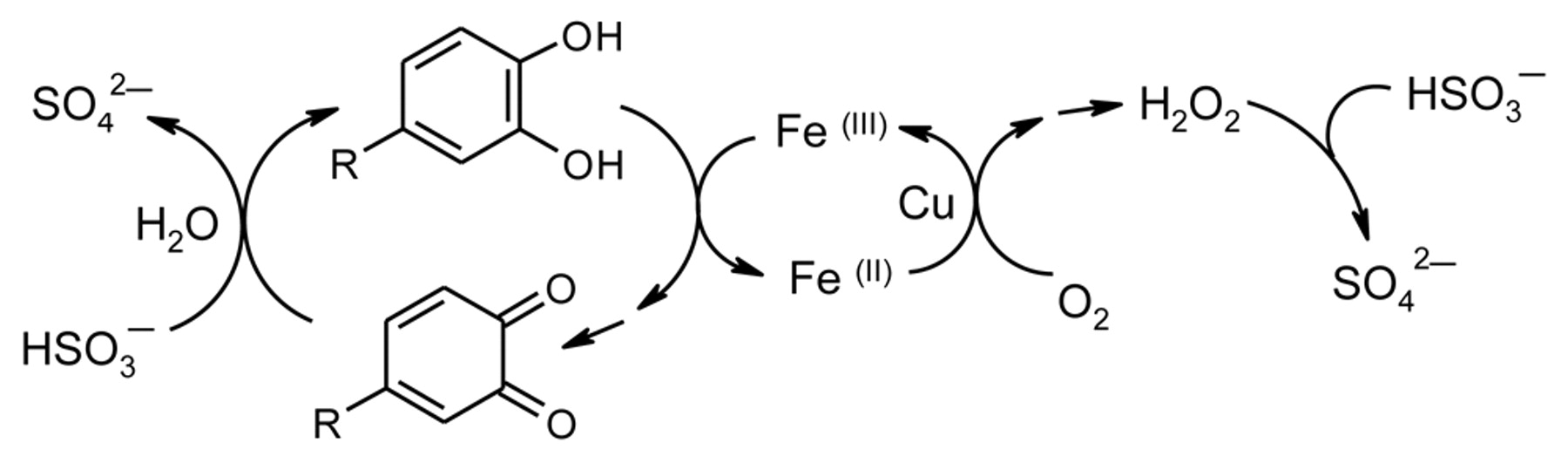
Proposed interaction of a catechol and O2 in the presence of sulfite.
In addition to protecting wine against the consequences of oxidation, sulfite accelerates the oxidation of polyphenols while also regenerating them from quinones. Alone, in the absence of sulfite or other nucleophile, catechols such as 4-MeC and (+)-catechin react with O2 extremely slowly in model wine despite the presence of metals; this accelerating effect of sulfite is also observed in wine (Danilewicz et al. 2008, Danilewicz and Wallbridge 2010). The goal of this work was to probe further into the mechanism of interaction of O2, polyphenols, and sulfite and the involvement of Fe and Cu. More specifically, the aims were to (1) confirm that there is an initial rapid O2 consumption associated with Fe(II) oxidation in model wine and wine before significant catechol autoxidation occurs, as described by Ribéreau-Gayon; (2) provide further evidence that catechols, such as (+)-catechin, in accord with reduction potentials, are oxidized extremely slowly in wine conditions and that rates of reaction with O2 are markedly accelerated by substances that are capable of trapping quinones; and (3) show that in the presence of sulfite the rate of reaction of O2 differs for different catechols to provide further evidence that O2 is not reacting with sulfite directly.
Materials and Methods
Materials.
Water (Fe ≤1 μg/L, Cu ≤0.4 μg/L; Emsure, Merck, Darmstadt, Germany), Cu(II) sulfate pentahydrate, Fe(III) chloride hexahydrate, sodium hydroxide, (+)-tartaric acid (BDH AnalaR grade), and ethanol (96% GRP grade) were obtained from VWR International (Lutherworth, UK). Benzenesulfinic acid sodium salt (BSAS), (+)-catechin, (−)-epicatechin, caffeic acid, gallic acid, and Fe(II) sulfate heptahydrate (99+% ACS reagent) were obtained from Sigma-Aldrich (Poole, Dorset, UK) and potassium metabisulfite (Kadifit) from Erbslöh Geisenheim AG (Geisenheim, Germany). Wine analyses were performed by Corkwise Ltd. (Nutfield, UK).
Wine.
The wine was a Reichensteiner (Riesling x Sylvaner x Madeleine Angevine cross), Canterbury Choice, Dry Reserve 2009 (Barnsole Vineyard, Canterbury, UK). Wine parameters were the following: 12.5% alc. (vol), pH 3.19, 6.7 g/L titratable acidity (as tartaric acid), 32 mg/L free SO2, 70 mg/L bound SO2, 1.7 mg/L Fe, and 0.08 mg/L Cu.
Preparation of model wine solutions.
For model wine solutions, 16.0 g (+)-tartaric acid was dissolved in water (~1.6 L) in a 2 L volumetric flask. Ethanol was added to give a 12% (v/v) final concentration. The pH was increased to 3.60 with 2.5 N sodium hydroxide, also adding water progressively to the mark as the required pH was approached. The concentration of tartaric acid was selected so that it would simulate the action of all the wine acids and produce a final titratable acidity of ~4 g/L.
Reaction of model wines with O2.
(a) Oxidation of polyphenols in the absence of sulfite.
Model wine was shaken in air until saturated with O2. It was then transferred to five 1 L volumetric flasks and (+)-catechin hydrate, (−)-epicatechin, gallic acid, caffeic acid, and pyrogallol (1.7 x 10−3 M) were added to separate flasks. Pyrogallol was readily soluble but the other polyphenols required ~4 hr to dissolve, except for caffeic acid, which required ~12 hr. Fe(II) (5 mg/L) and Cu(II) (0.3 mg/L) as aqueous solutions of their sulfates (250 μL and 100 μL, respectively) were added to each flask to initiate oxidation. Fourteen bottles were sealed immediately after the metal additions for each polyphenol, each set requiring ~10 min. Starting O2 concentration was measured in triplicate using the remaining solution immediately thereafter.
(b) Oxidation of (+)-catechin and increasing concentrations of Fe and Cu.
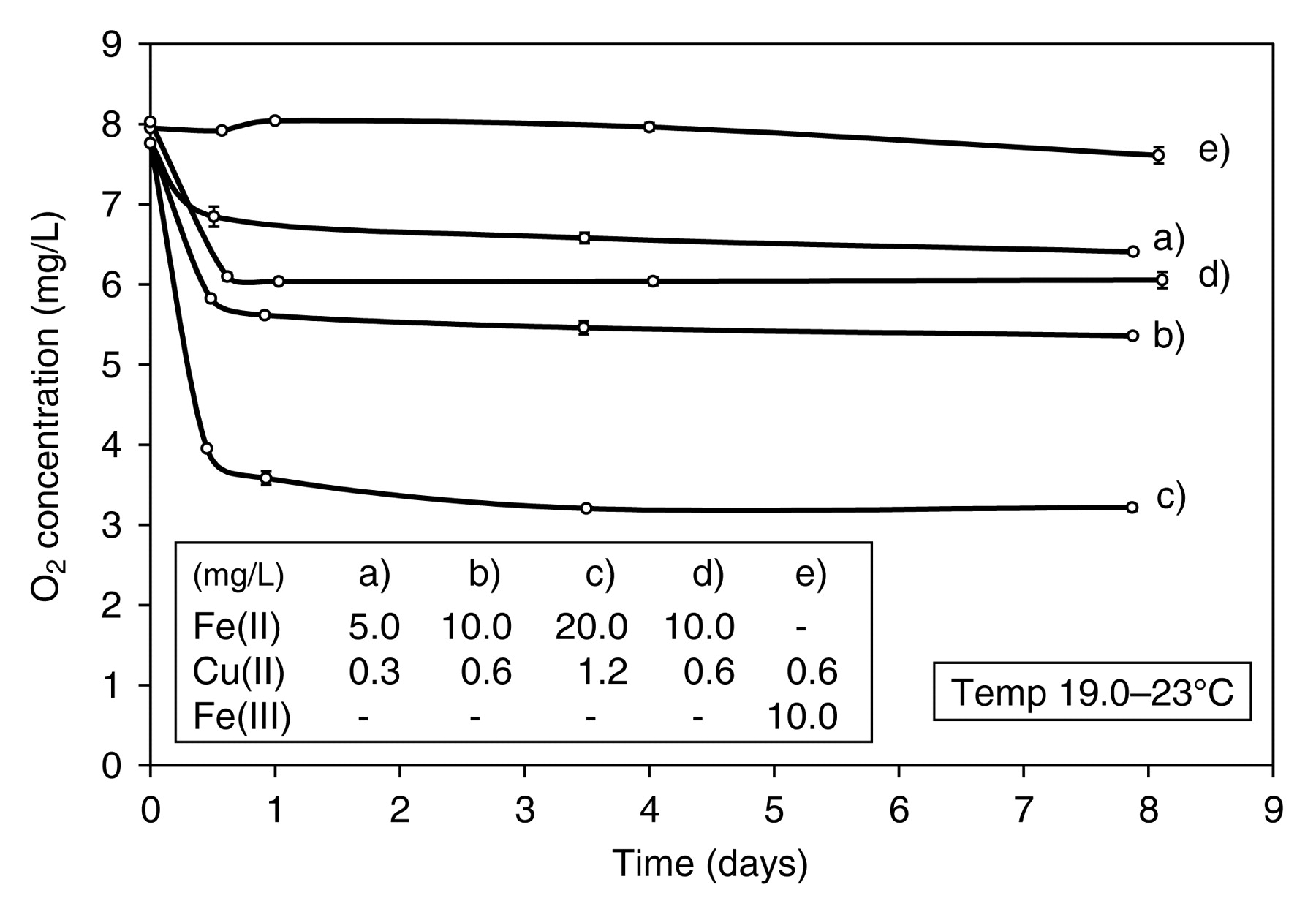
Model wine in which 524 mg/L (+)-catechin had been dissolved was saturated with air and poured into 1 L volumetric flasks. FeSO4.7H2O as a solid and CuSO4.5H2O as an aqueous solution (50, 100, and 200 μL in successive experiments) were added to each flask to give 5.0 mg/L Fe(II) and 0.3 mg/L Cu(II) in one flask, 10.0 mg/L Fe(II) and 0.6 mg/L Cu(II) in the second, and 20.0 mg/L Fe(II) and 1.2 mg/L Cu(II) in the third. In a subsequent experiment, FeCl3.6H2O was added as a solid to give 10 mg/L Fe(III) together with 0.6 mg/L Cu(II). In a further flask solid FeSO4.7H2O was added to give 10 mg/L Fe(II) together with 0.6 mg/L Cu(II), but without (+)-catechin present as a control. Bottles were then sealed as described above. When sulfite was present, it was added just after (+)-catechin addition. With model wine containing 10 mg/L Fe(II) and 0.6 mg/L Cu(II), one further set of bottles was sealed immediately after metal addition and a second set after 7.25 hr, maintaining O2 saturation to preoxidize the Fe(II).
(c) Effect of sulfite on the oxidation of pyrogallol.
Pyrogallol (214 mg/L, 1.7 x 10−3 M) was dissolved in 2 L of model wine, which had been saturated with air. Fe(II) (5 mg/L) and Cu(II) (0.3 mg/L) were added as described in (a) and 14 bottles were sealed within 10 min of this addition. Potassium metabisulfite (100 mg/L) was added to the remaining liter to give 52.8 mg/L SO2 and a second set of bottles was filled immediately. The rates of oxidation were followed with the two sets of bottles side by side.
(d) Effect of SO2, BSA, azide, and phloroglucinol on (+)-catechin oxidation.
Potassium metabisulfite (100 mg/L) was added to 1 L of air-saturated model wine in which (+)-catechin (524 mg/L, 1.7 x 10−3 M) had been dissolved as described in (a), giving 55.1 mg/L (8.4 x 10−4 M) free SO2. Benzenesulfinic acid sodium salt (BSAS) (1.93 g/L, 6.8 x 10−3 M) was added to a second 1 L of the above solution, NaN3 (442 mg/L, 6.8 x 10−3 M) to a third, and phloroglucinol (827 mg/L, 5.1 x 10−3 M) to a fourth. Fe(II) (5 mg/L) and Cu(II) (0.3 mg/L) were added as described in (a). Sets of bottles were sealed within ~10 min after each final metal addition and rates of O2 consumption were compared.
(e) Comparison of rates of oxidation of (+)-catechin, (−)-epicatechin, caffeic acid, and gallic acid (1.7 x 10−3 M) with sulfite.
Potassium metabisulfite was added to model wine containing 5.0 mg/L Fe(II) and 0.3 mg/L Cu(II) to give ~50 mg/L free SO2. This solution was air-saturated and left to stand for ~10 hr to oxidize the Fe(II) and then poured into four 1 L flasks. (+)-Catechin hydrate (524 mg), (−)-epicatechin (493 mg), caffeic acid (306 mg), and gallic acid (289 mg) were added to separate flasks. The (+)-catechin dissolved within 2 hr, but the other polyphenols required 6 to 11 hr to achieve complete dissolution. Since sulfite was present, any polyphenol that was oxidized during this time was presumed to have been largely regenerated. Each solution was then shaken in air to ensure saturation, and four sets of bottles were sealed and placed side by side to compare rates of oxidation. Initial free SO2 concentrations were also determined.
(f) Effect of pH and BSA on oxidation of (+)-catechin.
Model wine (2 L) containing 5.0 mg/L Fe(II) and 0.3 mg/L Cu(II) made up to pH 7.30 with 2.5 N sodium hydroxide was air-saturated. (+)-Catechin (524 mg/L), which dissolved in 25 min, was added to give a purple solution, which was sealed immediately in a set of 14 bottles, with a final pH 6.97. BSAS (1.64 g) was added to the remaining liter of model wine, which was sealed immediately in a further set of 14 bottles. The above procedure was repeated at pH 3.6, giving a further two sets of bottles. The four sets were placed side by side to compare the rates of O2 uptake.
Reaction of wine with O2.
The white wine (3 L) was shaken in air until saturated with O2 and free SO2 concentration raised to 49 mg/L by adding120 mg potassium metabisulfite. Fe(II) (8.3 mg/L) was added as solid FeSO4.7H2O (124 mg) followed by 0.22 mg/L Cu(II) as an aqueous solution of CuSO4.5H2O (250 μL). One set of 20 bottles was sealed within 10 min. The remaining solution was left for 10 hr to oxidize the added Fe(II), saturated with air, and a second set of bottles sealed.
O2 and SO2 measurement.
Solutions were sealed in ~60 mL brown glass bottles with screwcaps fitted with a plastic cone liner, ensuring the exclusion of any air bubbles and then stored in the dark. The small air space between the liner and the cap was filled with a polymeric filler to ensure that no O2 passed through the plastic cone into solutions during the course of experiments. Initial free SO2 concentration was determined in duplicate and O2 concentration in triplicate. Three bottles were taken at each time point to measure O2 concentrations in triplicate. Free SO2 was also measured when the O2:SO2 molar reaction ratio was determined. Mean values (±SD) were calculated and figures drawn using Excel software (Microsoft, Redmond, WA). (Where error bars denoting ±SD are not shown they were smaller than the data point symbol dimensions.) Maximum and minimum temperatures were noted during the course of each experiment.
An HI-9146 dissolved O2 meter fitted with a HI-76408 Clarke type electrode was used to measure O2 (Hanna Instruments, Leighton Buzzard, UK). The manufacturer specifies a resolution and limit of detection (LOD) of the meter as 0.01 mg/L O2. The bottle caps were quickly removed and a small stirrer bar inserted followed by the electrode, which displaced ~13 mL liquid. The electrode tip was lowered to ~5 mm of the briskly stirred magnetic bar. Readings stabilized within 30 sec and then remained stable for more than 5 min, showing that, although the system was not sealed during measurement, no measurable amount of external O2 reached the measurement area during that time.
Free SO2 concentration in model wine and white wine was measured with the modified Ripper procedure, using potassium iodate and starch-KI (Ough and Amerine 1988).
Results and Discussion
(+)-Catechin reacts with O2 extremely slowly in wine conditions in the absence of sulfite, despite the presence of Fe and Cu, as do (−)-epicatechin, gallic acid, and caffeic acid (Figure 1). After an O2 uptake of ~1.0 mg/L at the initial measurement point, presumably due to the oxidation of Fe(II), there appeared to be little further reaction. Fe(II) has been shown to oxidize rapidly in wine conditions and its rate of oxidation is markedly increased by Cu (Danilewicz and Wallbridge 2010). This rapid oxidation of Fe(II), first proposed by Ribéreau-Gayon (1931), becomes more apparent when O2 uptake is measured at earlier time points as with caffeic acid (Figure 1, curve d) and with (+)-catechin when exposed to increasing metal concentrations (Figure 2, curves a, b, c). After an initial uptake, which is approximately proportional to Fe/Cu concentration, essentially no further, or at least very little, oxidation of (+)-catechin (524 mg/L, 1.7 x 10−3 M) was observed even at very high metal concentrations. Sufficient (+)-catechin was present to react with ~54 mg/L O2 (~7 saturations of aerial O2). To confirm that the O2 uptake was due to the oxidation of Fe(II), the reaction was repeated with Fe(III) (Figure 2, curve e) and the rapid initial phase of O2 uptake was not evident. However, some slight uptake (0.34 ± 0.16 mg/L) appeared to occur over the eight days of the study at these high metal concentrations. When Fe(II) was oxidized in the absence of (+)-catechin, a similar rapid initial O2 uptake was observed as in its presence (Figure 2, curves d and b), but then no further reaction occurred. It can therefore be concluded that the initial O2 uptake was due to the reaction with Fe(II) and that any subsequent oxidation of (+)-catechin and the other three polyphenols is extremely slow.

Uptake of O2 by polyphenols (1.7 x 10−3 M) in model wine in the absence of sulfite; 5.0 mg/L Fe, 0.3 mg/L Cu.
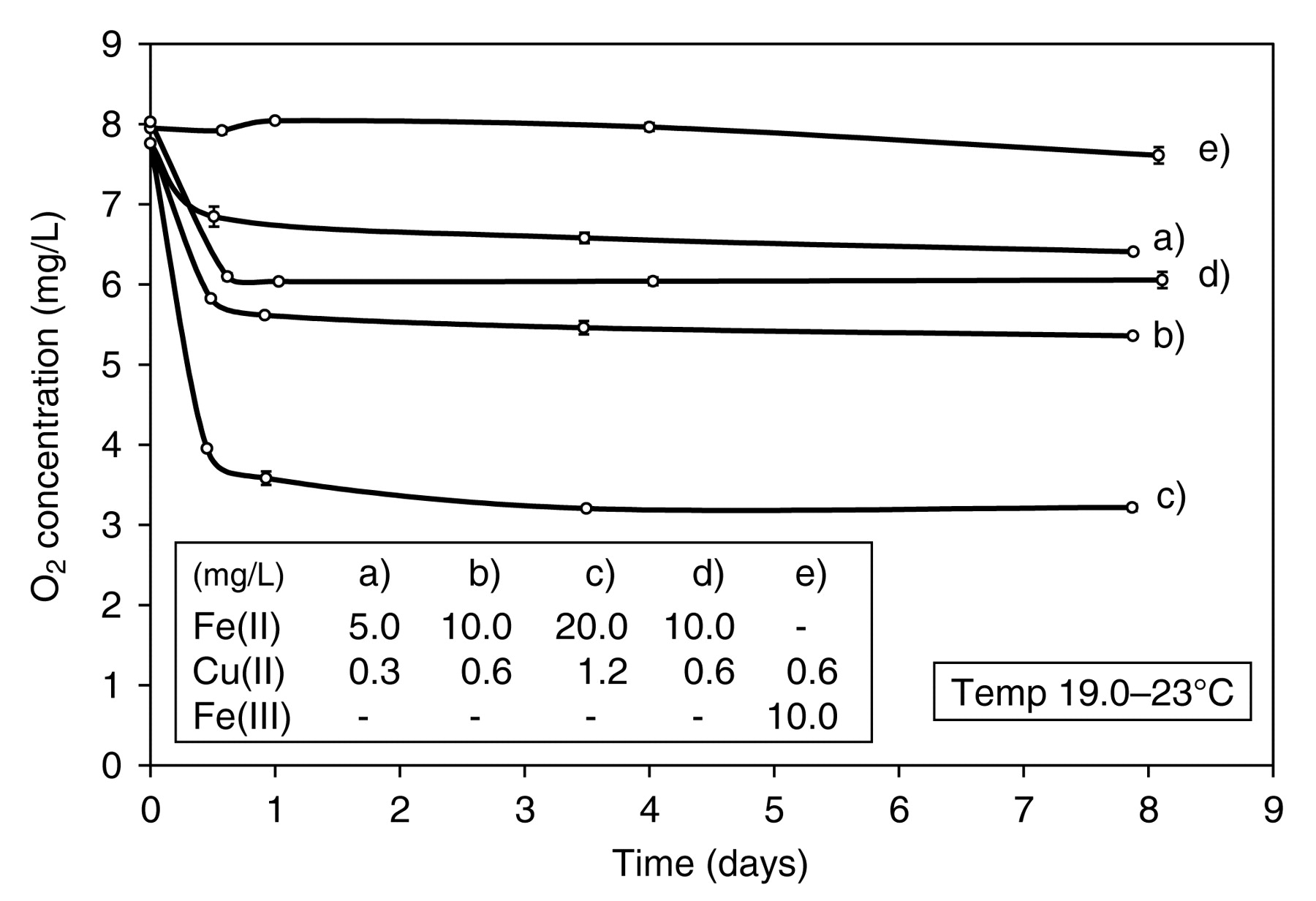
Oxidation of (+)-catechin (524 mg/L, 1.7 x 10−3 M) in model wine in the absence of sulfite. (a, b, c) Effect of increased Fe(II) concentration; (d) oxidation of Fe(II) without (+)-catechin present; (e) oxidation of (+)-catechin (524 mg/L, 1.7 x 10−3 M) in the presence of Fe(III).
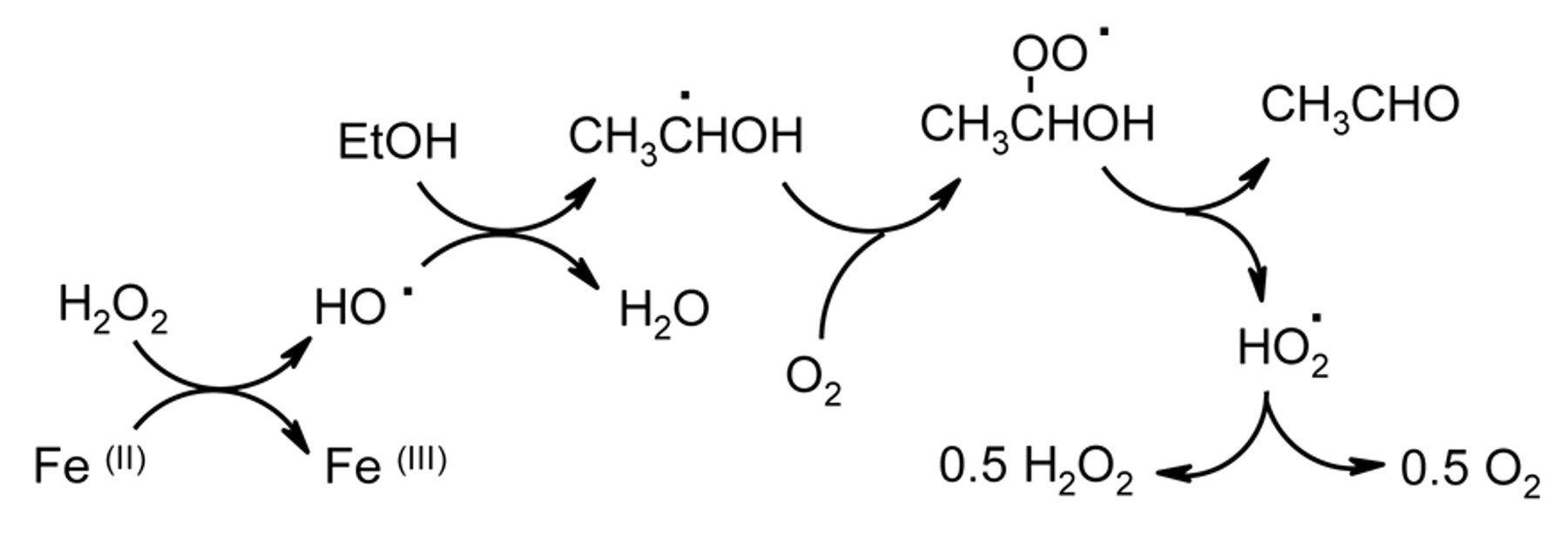
A detailed study of the Fenton reaction has shown that when Fe(II) is added to model wine containing an excess of hydrogen peroxide, the hydrogen peroxide produces an equimolar amount of acetaldehyde (Elias and Waterhouse 2010). The extent of reaction is not determined by the Fe(II) concentration, as the 1-hydroxyethyl radical, which is highly reducing, reduces the Fe(III) back to the ferrous state to continue its catalytic action (Scheme 2).

Fenton reaction in model wine in the absence of O2 and polyphenol.
However, when the reaction is repeated in the presence of air, the Fe(II) only produces an equimolar amount of acetaldehyde (Scheme 3) (Elias and Waterhouse 2010), as the 1-hydroxyethyl radical reacts with O2 to give a peroxyl radical and the acetaldehyde is produced by elimination of a hydroperoxyl radical (Bielski and Arudi 1983). Consequently, the Fe(III) is not reduced to continue the reaction and the amount of acetaldehyde formed is, therefore, dependent on the original Fe(II) concentration (Elias and Waterhouse 2010). Furthermore, since the Fe(II):acetaldehyde molar ratio is limited to 1:1, it also appears that Fe(II) does not reduce the hydroperoxyl radical and it is proposed that it disproportionates to yield O2 and hydrogen peroxide, which is highly favored thermodynamically (reaction 1) (Bielski and Arudi 1983).

Fenton reaction in model wine in the presence of O2 but in the absence of a polyphenol.
The Fenton reaction is used in the FOX-2 method for the determination of Fe(II) (Banerjee et al. 2002). Hydrogen peroxide, added in the presence of methanol as hydrogen donor oxidizes Fe(II) quantitatively to Fe(III), the concentration of which is then measured calorimetrically using the strong Fe(III) ligand, xylenol oranges. Here Fe(III) is stabilized and also not recycled back to the ferrous state in the presence of O2.
In the Fenton reaction involving O2 described above, 1.0 mole of Fe(II) is oxidized as 0.5 moles of O2 are consumed, 0.5 moles being regenerated (Scheme 3). It can also be assumed that two moles Fe(II) are required to reduce one of O2 to hydrogen peroxide. Therefore, when Fe(II) is oxidized in model wine in the absence of polyphenols, the Fe(II):O2 molar reaction ratio should be 2:1.
When a polyphenol is present it would react with the Fe(III) that is produced as Fe(II) is oxidized, returning it to the ferrous state to continue the catalytic process. It may also be oxidized by the hydroperoxyl radical (Danilewicz 2003, Waterhouse and Laurie 2006). From the initial results presented here, a polyphenol such as (+)-catechin, which is oxidized very slowly, does not appear to affect the Fe(II)/Fe(III) redox equilibrium significantly and it remains to be established how this equilibrium is maintained. The presence of a polyphenol should not interfere with the Fenton reaction. The HO• radical is a very powerful oxidant, which reacts at diffusion controlled rates. Therefore, it is proposed that it reacts close to its site of production with the first potential substrate it encounters. In the experiment here (Figure 2), ethanol is in 130-fold molar excess relative to (+)-catechin and so should be the most favored reactant, with tartaric acid, in 30-fold molar excess, second. The greater reducing ability of (+)-catechin should not enhance its reactivity. If reactivity depends on relative molar concentration, then the probability of an HO• encountering a (+)-catechin molecule would be ~0.5%.
Assuming a 2:1 molar reaction ratio, the initial 5 mg/L Fe(II) that was added should take up 1.4 mg/L O2 at most, and the observed ~1.0 mg/L uptake would therefore result in ~60% oxidized to Fe(III) (Figure 2, curve a). At 10 and 20 mg/L Fe(II) the O2 uptake at ~11 hr was 2.14 and 4.18 mg/L (curves b and c), which corresponds to ~74% of the Fe(II) being oxidized. After this initial uptake, the consumption of O2, which might be attributable to (+)-catechin, was barely perceptible. In the absence of (+)-catechin, 10 mg/L Fe(II) produced an uptake of 1.99 mg/L (curve d), corresponding to 69% oxidation. In two further experiments conducted side by side, the rates of O2 consumption by model wine containing Fe(II) (10 mg/L) and Cu(II) (0.6 mg/L) alone and in the presence of (+)-catechin (524 mg/L, 1.7 x 10−3 M) were compared in greater detail over 48 hr. The curves obtained for O2 consumption (data not shown) were almost superimposable, with uptake complete by 24 hr, most of which occurred by 6 hr. Total uptake after 48 hr without (+)-catechin was 2.16 ± 0.08 mg/L and with (+)-catechin was 2.29 ± 0.04 mg/L. Therefore, (+)-catechin appears to have little effect on the Fe(III):Fe(II) ratio at the initial equilibrium. Similar results were obtained in a previous study in which Fe(II) was oxidized in wine conditions and Fe(II) concentration followed using the ferrozine method. Equilibrium was then attained at 74% oxidation (Elias and Waterhouse 2010). However, Cu, which has a marked effect on the rate of Fe(II) oxidation (Danilewicz and Wallbridge 2010), was not included in this latter study, which probably explains why several days were required for equilibrium. In white wine, Fe remained as Fe(II) as long as it was protected from air; however, in wine containing 6 mg/L Fe(II), ~30% was oxidized after 24 hr on air exposure, as determined by measuring Fe(III) concentration (Ribéreau-Gayon 1931). Evidently, when model wine and wine is exposed to air not all the Fe(II) is oxidized. What controls this redox equilibrium, which appears to be little affected by polyphenols, is unclear.
Although (+)-catechin was oxidized extremely slowly, the two phases of oxidation were discernible when the oxidation of 4-MeC and pyrogallol was examined. Initial readings at 12 and 24 hr revealed that the rapid oxidation of Fe(II) was again apparent, followed by slower oxidation presumably of the phenols (Figure 3). The 4-MeC reduction potential (E3.6) is 36 mV lower than that of (+)-catechin, which explains its slightly faster rate of oxidation. Pyrogallol is rapidly oxidized (Figure 1, Figure 3), but earlier readings revealed the change in reaction rate at ~24 hr. Gallic acid may be oxidized at a slightly faster rate than the two flavanols (Figure 1), likely because of the relative instability of the corresponding quinone (discussed below).

Comparison of the uptake of O2 by (+)-catechin (524 mg/L), 4-methylcatechol (4-MeC) (211 mg/L), and pyrogallol (214 mg/L) (all 1.7 x 10−3 M) in model wine in the absence of sulfite; 10.0 mg/L Fe, 0.6 mg/L Cu; as for procedure (a) in the methods section.
The reduction potential of redox couples provides a means of comparing the strength of oxidizing and reducing agents; that is, their relative ability to accept or donate electrons and so to react with each other. The reduction potential of the O2/H2O2 redox couple (Scheme 4, half-reaction 2) at pH 7 (E7) is 365 mV for 1 M O2, with reference to the standard hydrogen electrode (Meisel and Czapski 1975, Wood 1988). Since protons are involved, the reduction potential is pH dependent so that

Reduction potentials of the O2/H2O2 and quinone/catechol couples at pH 3.6. Combination of the two ‘half’ reactions in the oxidation of (+)-catechin.
where EpH is the reduction potential at a required pH and E0 is the reduction potential at the standard state (pH 0). Therefore, the potential falls by 59 mV for each unit increase in pH (Meisel and Czapski 1975, Kilmartin et al 2001) and, consequently, E0 = 778 mV and E3.6 = 566 mV for the above redox couple.
Similarly, the quinone/catechol redox couple may be represented in half-reaction 3 (Scheme 4). The reduction potential will vary depending on group R, but for (+)-catechin, E3.6 is 577 mV in a wine model system by cyclic voltammetry (Table 1) (Kilmartin et al. 2001). The two half-reactions 2 and 3 may be combined by reversing the latter to give reaction 4. The difference in reduction potential (ΔE) is close to zero and from the relationship ΔE = 0.029logK for a two-electron process at 25°C, the equilibrium constant, K, will be 1. A positive ΔE value would favor reaction to the right and a negative value to the left. For simple catechols, this reaction should not proceed to completion (Danilewicz 2003). From the relationship ΔG = −nFΔE, where n is the number of electrons transferred and F = Faraday constant, there would be no change in Gibbs free energy (ΔG) (Atkins 1999), and it is found, despite the presence of large concentrations of metals, that (+)-catechin reacts with O2 very slowly in wine conditions.
Reduction potentials of quinone/catechol redox couples.
The reduction potential of the corresponding redox couples of (−)-epicatechin, caffeic acid, and gallic acid are similar to that of (+)-catechin in winelike conditions (Table 1) (Kilmartin et al. 2001). The corresponding calculated E0 values for caffeic acid and quercetin agree reasonably well with values derived from potentials measured at pH 6.2 in phosphate buffer (Horner and Geyer 1965). In this latter study, E0 for the pyrogallol couple was 680 mV, which gives a corresponding E3.6 of 468 mV. Pyrogallol is clearly a stronger reductant than the above polyphenols. ΔE3.6 for reaction 5 (Scheme 5) now becomes 98 mV, which results in an equilibrium constant K = 2.4 x 103 and a ΔG of −18.9 kJmol−1. Thus, it is readily oxidized in wine conditions (Figure 1, Figure 3).

Oxidation of pyrogallol.
Comparing the reduction potential of the 1,2-benzenediol and (+)-catechin couples, the tetrahydrochroman system in the latter compound reduces the potential slightly (5 mV). Therefore, based on the value for pyrogallol, E3.6 for (+)-gallocatechin and (−)-epigallocatechin should be ~463 mV. These substances and tannins containing them will therefore be prime oxidation targets in the absence of sulfite. In contrast, the reduction potential of the pyrogallol system in gallic acid and its esters, such as (−)-epicatechin gallate, is raised considerably by the electron-withdrawing effect of the carboxylate function, which accounts for its much reduced susceptibility to oxidation (Danilewicz 2003).
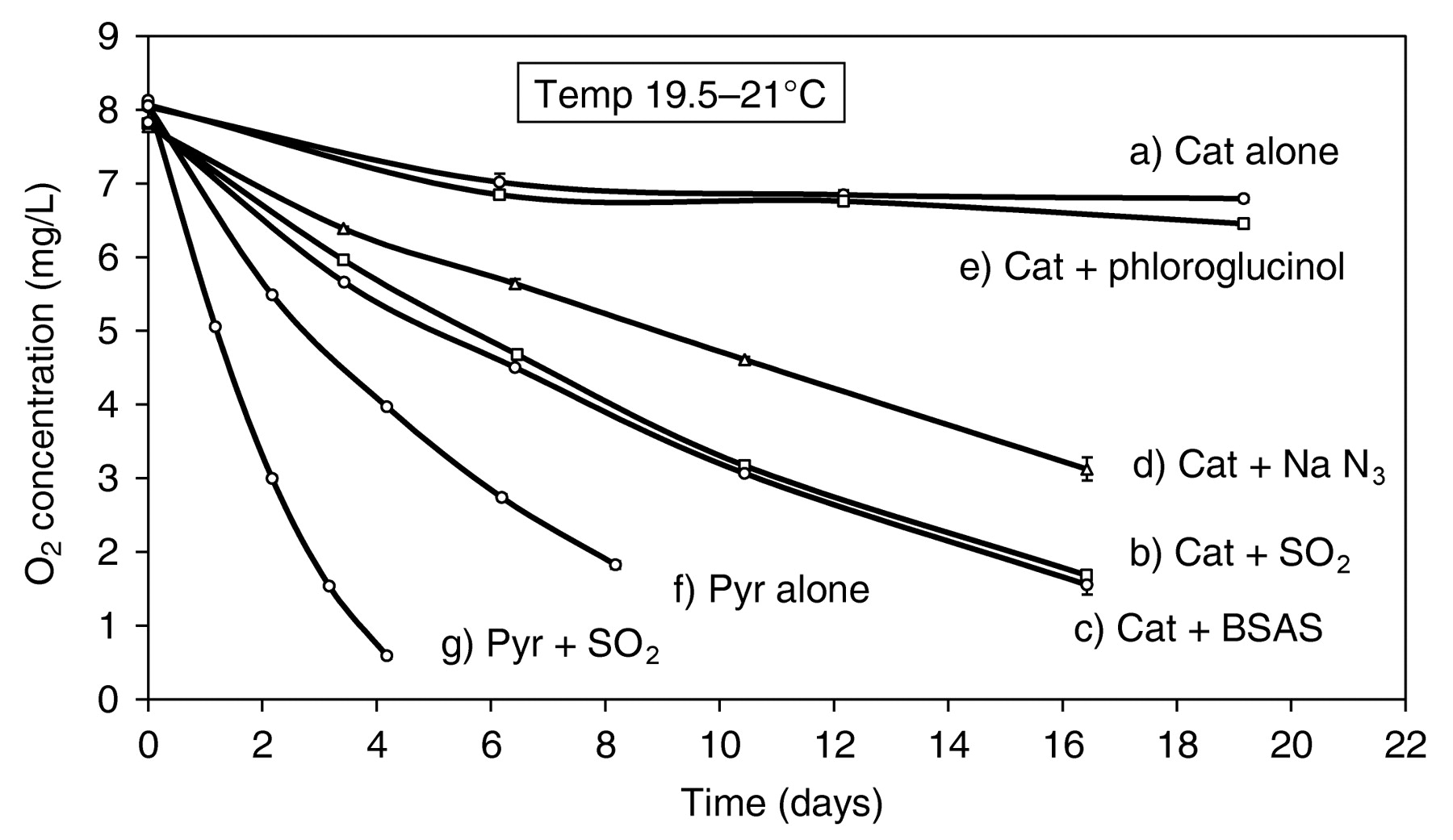
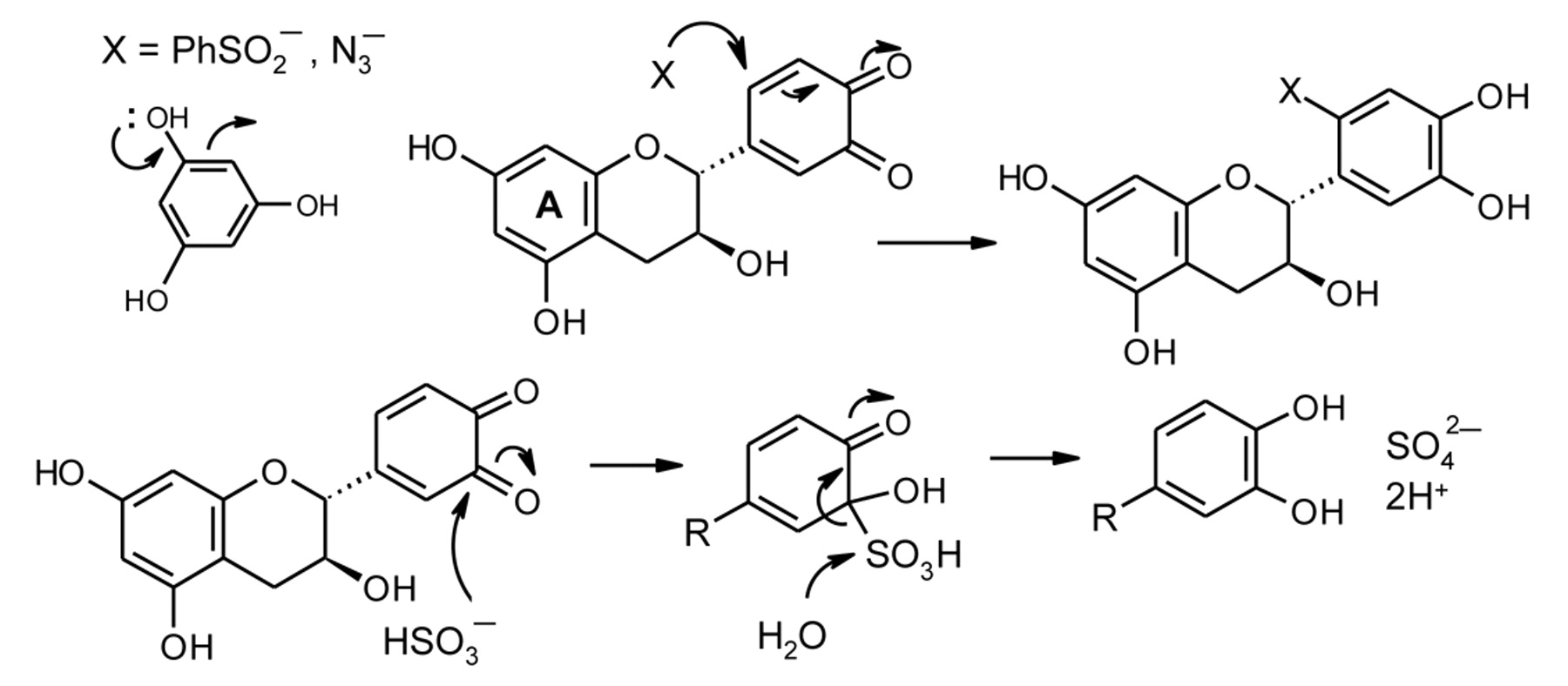
Even though (+)-catechin is not oxidized at a significant rate in model wine, addition of sulfite results in prompt O2 uptake (Figure 4, curves a and b). There is also an augmentation in the rate of O2 consumption with pyrogallol (curves f and g). Sulfite reacts rapidly with quinones and it has been proposed that sulfite, by intercepting them, displaces the reversible slow redox process to drive the oxidation forward (Danilewicz and Wallbridge 2010). This conclusion was supported by the observation that BSA, which does not react with O2 in model wine conditions, also accelerates the reaction (curve c). BSA is known to react with the (+)-catechin quinone rapidly by 1,4-conjugate addition (Scheme 6) (Dangles et al. 2000), and the adduct was formed when (+)-catechin was oxidized in model wine in the presence of BSA (Danilewicz and Wallbridge 2010). This proposal has now been further supported by the finding that azide, which also reacts in this manner with unsaturated systems, including quinones (Cassis et al. 1987, Gauthier et al. 2001), accelerates the rate of O2 consumption (Figure 4, curve d). Azide also does not react with O2 and, furthermore, cannot change its oxidation state. Unlike 4-MeC no evidence of a sulfonic acid adduct was observed by HPLC when (+)-catechin was oxidized in the presence of sulfite; the catechol is regenerated in high yield (Danilewicz and Wallbridge 2010). It is proposed that this mechanistic difference is due to increased steric hindrance to conjugate addition and the ability of sulfite preferentially to undergo reversible 1,2-addition, the adduct then eliminating sulfate to produce the catechol (Scheme 6). The ability of a substance to trap quinones is a factor in determining the rate of reaction of polyphenols with O2. In the azide reaction, the adduct must be unstable as the solution darkened rapidly, forming a black precipitate.

Uptake of O2 by polyphenols (1.7 x 10−3 M) in model wine in the presence of sulfite and other nucleophiles; 5.0 mg/L Fe; 0.3 mg/L Cu. (a) (+)-Catechin (Cat) alone; (b) Cat + SO2 free 53.3 mg/L, 8.3 x 10−4M; (c) Cat + benzenesulfinic acid sodium salt (BSAS) 1.116 g/L, 6.3 x 10−3 M; (d) Cat + NaN3 442 mg/L, 6.8 x 10−3 M; (e) Cat + phloroglucinol 827 mg/L, 5.1 x 10−3 M; (f) pyrogallol (Pyr) alone; (g) Pyr + SO2 free 52.8 mg/L, 8.25 x 10−4 M.
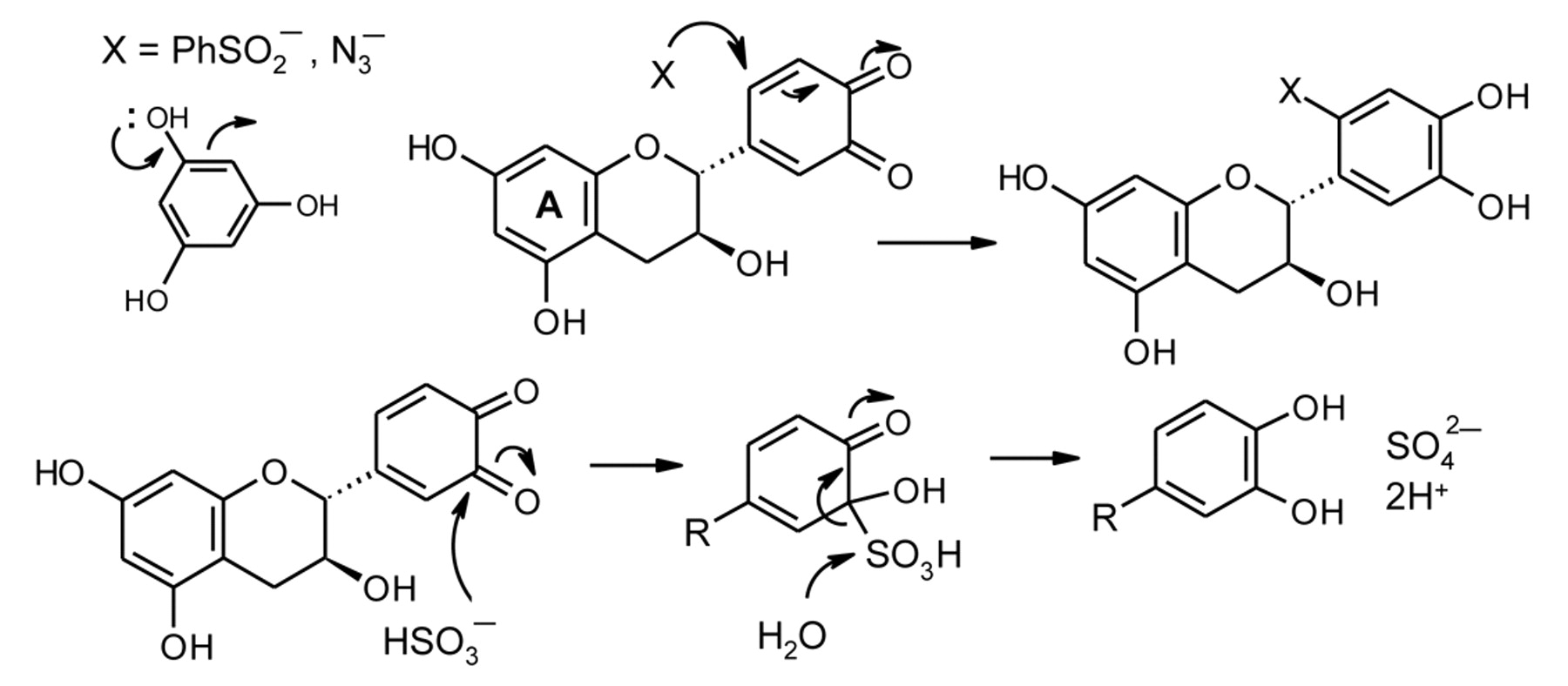
Conjugate addition of nucleophiles to (+)-catechin quinone and proposed contrasting carbonyl addition of sulfite to regenerate the catechol.
The proposed mechanism is similar to that proposed for the oxidation of caftaric acid by polyphenoloxidase (PPO), which is markedly accelerated by glutathione (Cheynier and Van Hulst 1988). The authors concluded that PPO oxidation of caftaric acid is extremely fast but that reduction of the quinone back to caftaric occurs simultaneously. Acceleration of this reversible redox process then resulted, as the quinone was trapped by reaction with glutathione, so preventing the reverse reduction. As with (+)-catechin, the reduction potential of the caftaric acid and O2 redox couples are very close and so the reaction cannot go to completion, as it is limited by the reverse reaction at equilibrium. The thermodynamics of the reaction still apply despite faster kinetics. PPO allows equilibrium conditions to be rapidly established.
In contrast, phloroglucinol, which can be taken as a model for the flavanol A-ring, does not appear to react with the quinone significantly as it has only a slight effect on the rate of oxidation. Consequently, dimerization of flavanols by this mechanism should not be a significant process in wine conditions (Guyot et al. 1996) and would be prevented by sulfite, which adds to the quinone much more rapidly at lower concentration. Furthermore, since (+)-catechin is oxidized no more rapidly than caffeic acid in the absence of sulfite, it does not promote its own oxidation by the interaction of its A-ring with its quinone.
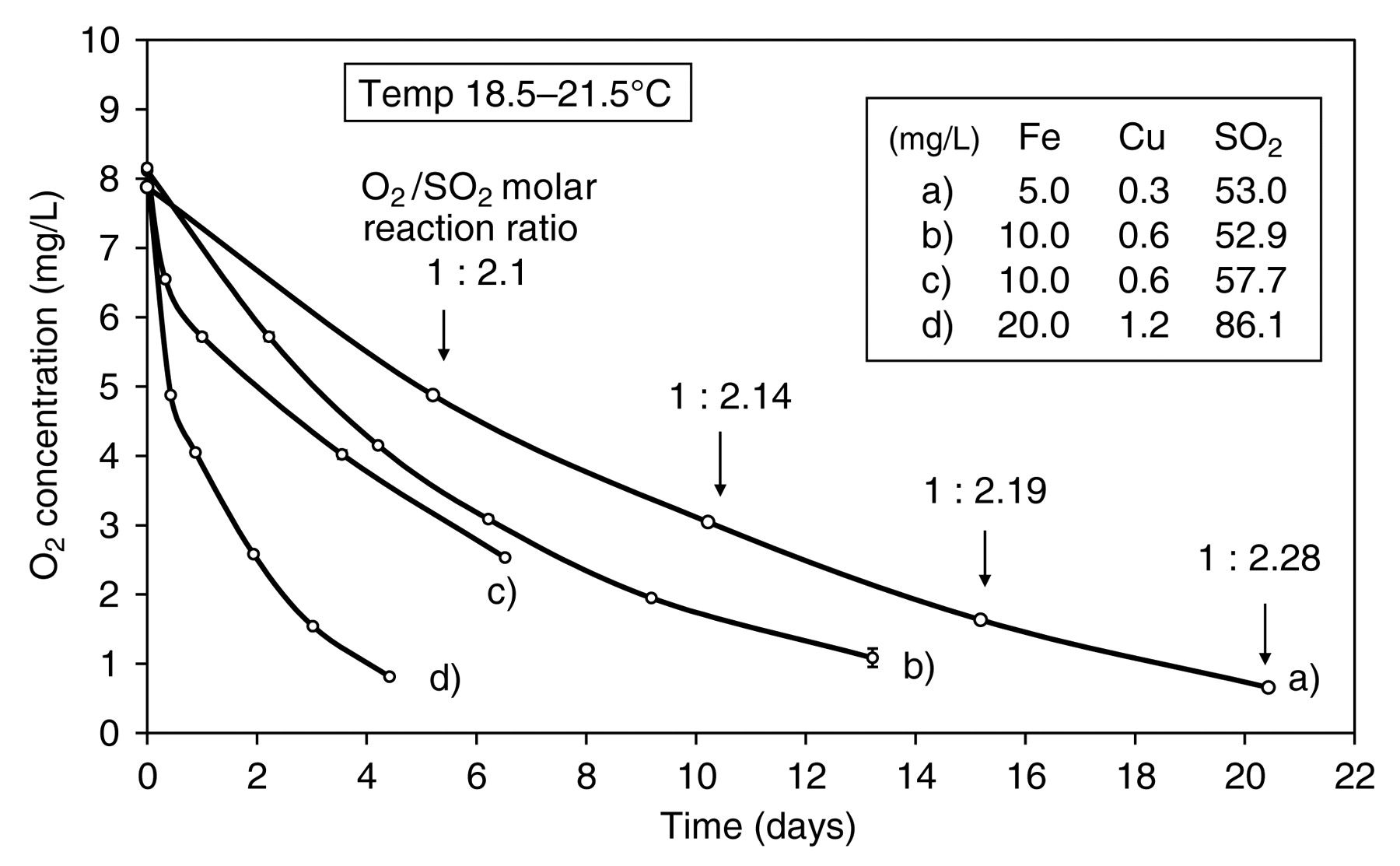
When (+)-catechin was oxidized in a closed system, sealing the sample bottles immediately after saturation with air and metal addition within a wine concentration range, a smooth uptake of O2 was observed. The O2:SO2 molar reaction ratio was close to 1:2 but tended to rise slightly as O2 was consumed (Figure 5, curve a). However, when the first determinations were made at time points earlier than three days, ratios as low as 1:1.6 were observed. This change in molar reaction ratio suggested that there is an underlying change in mechanism as oxidation proceeds. The cause of this change became apparent when the effect of larger concentrations of metals was examined. With 10.0 mg/L Fe(II) and 0.6 mg/L Cu(II), a rapid initial uptake of 1.6 mg/L O2 was observed after 8 hr with an O2:SO2 molar reaction ratio of 1:1.4, followed by smooth exponential decline in O2 concentration (Figure 5, curve c). When metal concentration was doubled again, the initial O2 uptake was 3.24 mg/L after 10 hr with a reaction ratio of 1:1.05 (curve d). This initial phase, when O2 is more rapidly consumed, appears to be due to oxidation of Fe(II), when sulfite reacts principally with hydrogen peroxide. Subsequently, the molar reaction ratio would increase, as sulfite also reacted with quinone as the polyphenol started to oxidize. Similarly, sulfite autoxidation in the presence of Fe(II) is characterized by an induction period when Fe(II) is first oxidized to Fe(III), which by oxidizing sulfite to the sulfite radical initiates oxidation (Huss et al. 1982). The remarkable effect of sulfite on the rate of oxidation of (+)-catechin is apparent when comparing Figures 2 and 5.

Oxidation of (+)-catechin (524 mg/L) with increased concentration of Fe and Cu in the presence of sulfite. Samples sealed immediately after metal addition, except for (b), where samples were sealed after Fe(II) oxidation.
However, Ribéreau-Gayon (1931) estimated that the O2 uptake was too great to be taken up entirely for the oxidation of Fe(II) to the ferric state, and he proposed that some of this initial O2 uptake was due to the direct reaction of O2 with wine constituents to produce intermediate labile oxidants, which he further proposed were peroxides, as described above. These calculations, however, appear to have underestimated the amount of Fe(II) that O2 would oxidize, despite not taking into account the effect of sulfite. Fe(II) would not be required to provide the four electrons to reduce O2 to water because the intermediate hydrogen peroxide would be intercepted by sulfite (Scheme 7). The reaction of sulfite with O2 would be suppressed in wine conditions, since sulfite autoxidation is a radical chain reaction that is inhibited by radical scavengers such as catechols (Danilewicz 2007). Therefore, only two electrons would be needed, and thus 10 mg/L and 20 mg/L Fe(II) would react with 2.86 mg/L and 5.73 mg/L O2, respectively. At the initial time points (Figure 5, curves c and d), the actual O2 uptake was 1.6 and 3.24 mg/L and so not all the Fe(II) was oxidized. Consequently, all the initial O2 uptake is accounted for, as discussed above when sulfite is absent, and there is no need to invoke the formation of peroxides (Ribéreau-Gayon et al. 2000), which in any case is unlikely as O2 would not react directly with any wine constituent other than Fe(II) at the observed rates. Hydrogen peroxide is the only peroxide formed and that is removed by sulfite.

Initial oxidation of Fe(II) in the presence of sulfite. O2:SO2 molar reaction ratio would be 1:1, with two Fe(II) oxidized for each O2 reduced before the initiation of polyphenol oxidation.
The change in O2:SO2 molar reaction ratio is therefore explained by the fast initial Fe(II) oxidation, but the overall ratio should remain ~1:2 when all the Fe returns to the ferrous state as O2 concentration approaches zero. When the oxidation of (+)-catechin in model wine containing 10 mg/L Fe(II) and 0.6 mg/L Cu(II) was repeated, sealing the samples 7 hr after the metals were added to allow most of the Fe(II) to be oxidized, and then resaturating with air, a smooth exponential decline in O2 concentration occurred (Figure 5, curve b). Unexpectedly for such a complex reaction, the uptake of O2 follows a first-order rate law in O2 (Equations 1 to 3), where [O2]0 is initial O2 concentration and [O2] is O2 concentration at time t (Pilling and Seakins 1995). Accordingly, a plot of ln[O2]/[O2]0 against time gives a straight line of slope k (0.15 days−1), which is the pseudo-first-order rate constant (Figure 6, curve a). This kinetic profile contrasts with that obtained when the model wine samples were sealed immediately after metal addition, when initial Fe oxidation was superimposed on that of (+)-catechin (Figure 6, curve b). The rate of oxidation of polyphenols is also dependent on their concentration (Danilewicz 2007). However, in these reactions (+)-catechin concentration should not change as it is oxidized, since sulfite regenerates it from the quinone, so allowing pseudo-first-order kinetics.

Plot of ln[O2]/[O2]0 against time from Figure 5; 10.0 mg/L Fe, 0.6 mg/L Cu. (a) Samples sealed after Fe(II) was preoxidized; (b) samples sealed immediately after metal addition.
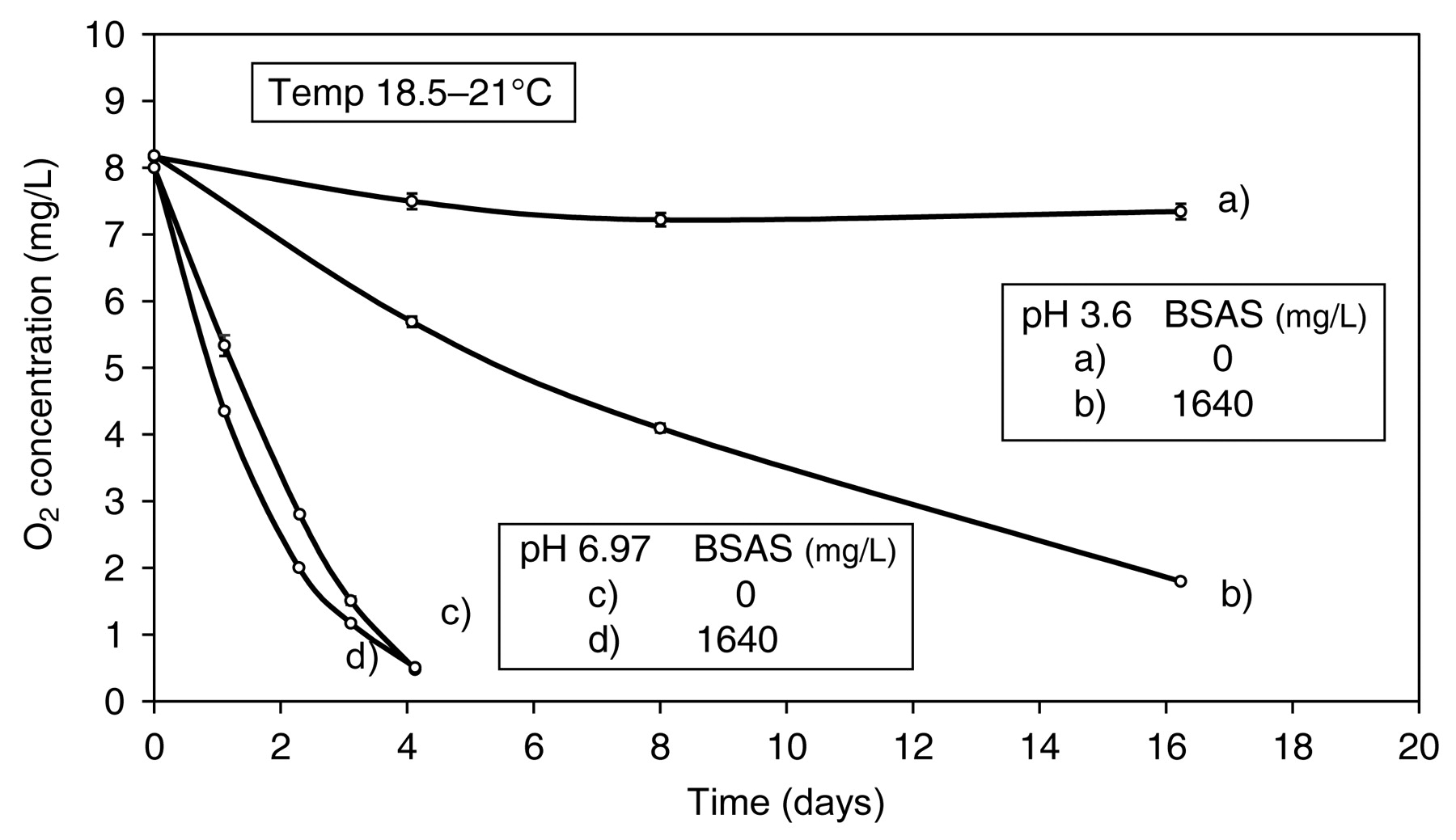
The rate of oxidation of polyphenols accelerates with increasing pH (Cilliers and Singleton 1989,1991). However, the reduction potentials of the O2/H2O2 and quinone/polyphenol redox couples have the same pH dependency, each reduced by 59 mV per unit increase in pH (Meisel and Czapski 1975). Therefore, ΔE for reaction 4 would remain close to zero as pH is increased and the reaction is no more favored thermodynamically at pH 9 than it is at pH 3. It is proposed that the increase in rate of oxidation (Figure 7) is due to the much-reduced stability of the quinone, which by decomposing with increasing rapidity as pH increases draws the reaction forward by displacing the redox process. While BSA allows (+)-catechin to react with O2 at pH 3.6 by adding to the quinone, at pH 6.97 its effect on reaction rate is much reduced. It is proposed that the rate of quinone decomposition is then comparable to the rate of BSA addition (Figure 7).

Effect of pH (3.6 and 6.97) on rate of oxidation of (+)-catechin (524 mg/L) in model wine, 5.0 mg/L Fe; 0.3 mg/L Cu, ± benzenesulfinic acid sodium salt (BSAS).
The rates of autoxidation of four polyphenols were compared at a concentration of 1.7 x 10−3 mol/L in the presence of sulfite, first allowing the Fe(II) to oxidize (Figure 8). The rate of reaction increased in the order of (+)-catechin < caffeic acid < (−)-epicatechin < gallic acid, despite the redox couples of the four polyphenols having similar reduction potentials. It is therefore proposed that the different rates of reaction are due to different rates of removal of the respective quinones. It has been observed that the quinone of (−)-epicatechin is less stable than that of (+)-catechin (Richard-Forget et al. 1992). A possibility is that the rate of decomposition of (−)-epicatechin quinone is fast enough to compete with its reaction with sulfite. The quinone would then be removed more quickly to accelerate oxidation. It has been observed that in the presence of sulfite, only ~79% of the quinone was reduced back to the catechol for (−)-epicatechin compared to ~96% for (+)-catechin (Danilewicz and Wallbridge 2010). Gallic acid is oxidized fastest both without and with sulfite (Figure 1, Figure 8) and its quinone is the most unstable as observed in cyclic voltammetry by loss of the return cathodic peak (Kilmartin et al. 2001). Perhaps the rates of oxidation of (+)-catechin and (−)-epicatechin are too slow to show a difference over 24 days in the absence of sulfite. For (+)-catechin and caffeic acid, O2 concentration declines exponentially. Therefore, for these polyphenols the rate of oxidation followed a pseudo-first-order rate law with respect to O2 and a plot of ln[O2]/[O2]0 gave straight lines, with rate constant k = 0.093 and 0.115 days−1, respectively (Figure 9). However, (−)-epicatechin and particularly gallic acid (curves c and d) deviate from linearity, indicating departure from first-order kinetics and the intrusion of an addition process. It is possible, therefore, that the concentration of caffeic acid, like that of (+)-catechin, does not diminish as it is oxidized in these experiments due to its regeneration by sulfite from the quinone.

Oxidation of polyphenols (1.7 x 10−3 M) in model wine in the presence of SO2; 5.0 mg/L Fe; 0.3 mg/L Cu.

Oxidation of polyphenols (1.7 x 10−3 M) in model wine. Plot of ln[O2]/[O2]0 against time from Figure 8.
In the kinetics of polyphenol oxidation, Fe is first oxidized. This initial activation of the catalyst is also observed in a white wine. The Reichensteiner wine, which had a low Cu concentration (Fe 1.7 mg/L, Cu 0.08 mg/L) was first maintained at O2 saturation overnight to allow the Fe(II)/Fe(III) to equilibrate. Fe(II) (8.3 mg/L) and Cu(II) (0.22 mg/L) were then added and a portion was sealed immediately, ensuring full O2 saturation. A slowing in rate was apparent at ~20 hr (Figure 10, curve a). When the wine was left to oxidize for 12 hr to allow the added Fe(II) to oxidize before the samples were sealed, a smooth curve was obtained with close to an exponential decline in O2 concentration (curve b). Consequently, a plot of ln[O2]/[O2]0 gave a straight line, with a rate constant of 0.139 days−1. This first-order rate law with respect to O2 would be anticipated based on the above result with caffeic acid, as it and derivatives are the main polyphenols in white wine.

Oxidation of a white wine: (a) samples sealed immediately after metal addition; (b) samples sealed after Fe(II) was preoxidized; 10.0 mg/L Fe; 0.3 mg/L Cu.
In a study of polyphenol oxidation, (+)-catechin, caffeic acid, and gallic acid were oxidized without the addition of Fe or sulfite (Wildenradt and Singleton 1974). It is therefore proposed that the reactions here were only possible due to the presence of trace metal impurities and the use of pure O2 to force the reactions. The temperature was also raised to 50°C, which would reduce the stability of the quinones.
In an autoxidation study in which grape seed polyphenols were separated into crude fractions containing flavanol monomers, procyanidin dimers and trimmers, and gallate esters, the rate of degradation increased in the order (+)-catechin < (−)-epicatechin < procyanidins and (−)-epicatechin degraded more slowly than its gallate ester in model wine (de Freitas et al. 1998). Although (+)-gallocatechin and (−)-epigallocatechin may have been present in some fractions, the reduction potential of most of the redox couples in these compounds should have been similar, and therefore it is proposed that the rates of degradation could be dependent on the stability of the respective quinones. However, the rates of reaction of (+)-catechin and (−)-epicatechin were much faster than those observed in this current study (Figure 1). Although higher metal concentrations were used (10.0 mg/L Fe and 1.0 mg/L Cu), the marked difference in rates could also be due to the presence of additional substances in the crude fractions that reacted with the quinones, since sulfite was not reported to be present. It seems that the rate of oxidation of a polyphenol does not depend solely on the reduction potential of its redox couple.
Conclusions
It is concluded that the oxidation of polyphenols depends on Fe redox cycling. After extended protection from air, when all the Fe is in the ferrous state, exposure of wine containing sulfite to O2 results in the initial oxidation of Fe(II), until a Fe(III)/Fe(II) redox equilibrium is attained. The factors that control this equilibrium, which appears little affected by polyphenols in the absence of sulfite, remain to be identified. In parallel, the rate of oxidation of polyphenols by Fe(III) should accelerate as Fe(III) concentration increases and then progressively decline as Fe(III) returns to the ferrous state with the consumption of O2. Over time, polyphenols eventually return the Fe(III) back to the ferrous state.
Although metals are essential catalysts for the oxidation of polyphenols in winelike systems, they alone do not control the rate of oxidation of simple polyphenols such as (+)-catechin, caffeic acid, and gallic acid with reduction potentials close to that of the O2/H2O2 redox couple. The presence of substances such as sulfite and other nucleophiles, capable of reacting with their quinones, allows oxidation to proceed at a much faster rate. However, once sulfite is present the rate of oxidation becomes highly dependent on metal concentration.
In the presence of sulfite, the oxidation of (+)-catechin obeys a first-order rate law in O2. It had been previously observed that, although sulfite accelerates polyphenol oxidation, paradoxically the rate of sulfite reaction, and hence the rate of oxidation, is largely independent of SO2 concentration. The kinetics of SO2 autoxidation are different, with the rate of SO2 oxidation dependent on its concentration but independent of that of O2. Sulfite autoxidation is dependent on the rate of formation of the sulfite radical, with which O2 reacts rapidly, such that this reaction does not influence overall reaction rate. This clear difference in mechanism supports the contention that sulfite does not react directly with O2 in wine.
- Received September 1, 2010.
- Revision received March 1, 2011.
- Accepted April 1, 2011.
- Published online September 2011
- © 2011 by the American Society for Enology and Viticulture
Literature Cited
Vol 62 Issue 3








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
More from this TOC section
Similar Articles
