


Abstract
Data from 13 years (78 wines) of wine industry laboratory proficiency testing were reviewed. After outlier removal, within-laboratory precision (repeatability) and across-laboratory precision (reproducibility) were determined for measurements of alcohol, titratable acidity, volatile acidity, total SO2, free SO2, malic acid, specific gravity, pH, residual sugar, glucose plus fructose, and absorbance at 420 and 520 nm. Reproducibility was 3.6 to 57.8 times higher than repeatability. Reproducibility was evaluated with Horwitz ratios (HorRat); only alcohol, titratable acid, and total SO2 had acceptable values (mean HorRat <2). Measurement z scores demonstrated non-normal distributions, particularly for specific gravity, likely due to confounding with density. Reproducibility did not vary significantly over time, with exceptions: imprecision of ethanol measurements decreased (improved) by 0.0084% v/v per year, while the imprecision of titratable acidity, pH, and malic acid measurements increased by 0.0089 g/L as tartaric, 0.0008 pH units, and 0.13 g malic acid/L per year, respectively. The imprecision of reproducibility and repeatability generally increased with analyte concentration, with notable exceptions for alcohol (both), volatile acidity (reproducibility), and total SO2 (repeatability). The methods or instruments used to determine alcohol, titratable acidity, free and total SO2, and volatile acidity changed significantly over time. Significant differences were observed among techniques for many analytes, which can be rationalized by attribution to well-known matrix effects manageable in a properly run method; e.g., higher apparent concentrations of alcohol by boiling point methods in high-sugar matrices. Evaluation of method accuracy was not possible due to the lack of wine reference materials with known true values. Results demonstrate the need for industry-wide improvement in analytical performance for some assays, and the potential benefit of adopting criteria guidelines for method performance.
- wine analysis
- winery laboratory
- proficiency testing
- performance criteria
- method validation
- wine quality
- HorRat
Chemical analysis in the wine industry
Winery laboratories run analysis to comply with regulations and to improve or ensure product quality (Amerine and Ough 1980). In the United States, the Department of the Treasury’s Alcohol and Tobacco Tax and Trade Bureau (TTB) requires that alcohol, total SO2, and volatile acid concentrations are within specified limits (Federal Alcohol Administration Act of 1935, Internal Revenue Code of 1986a), and specific gravity may be run as a means to check bottle fill level (Jacobson 2006). However, most routine wine analyses are performed to evaluate wine quality by measuring compounds associated with spoilage, stability, or sensory properties (Amerine and Ough 1980). These parameters are measured using analytical methods identical to or derived from published methods in the AOAC official methods (AOAC 2012), many of which are described in popular wine analysis texts (Amerine and Ough 1980, Iland et al. 2004, Jacobson 2006, Zoecklein et al. 1994). The TTB recommends but does not require that wineries use either AOAC methods or the methods used by its own laboratory (Alcohol and Tobacco Tax and Trade Bureau 2010), and the wine industry has thus operated under the “results-driven” or “fitness for purpose” principle rather than on dictated methods. “Fitness for purpose” requires that a method has accuracy and precision appropriate to the application. One method for determining precision is to test similar samples across multiple labs to evaluate both across-method and across-laboratory errors (Garfield et al. 2000, Wernimont and Spendley 1985). Collaborative testing also offers an opportunity to evaluate the analytical proficiency of a laboratory or individual regardless of the method used (ISO 2005), which is a necessary aspect of a multi-faceted laboratory quality program (Butzke and Ebeler 1999).
Analytical performance terminology
Several authors discuss the terminology of analytical performance (Butzke and Ebeler 1999, Garfield et al. 2000, Horwitz and Albert 2006). Collecting data to evaluate the analytical performance of individuals, laboratories, methods, or equipment is referred to as validation. Validation studies can potentially provide information about accuracy (closeness to the true value), precision (expected and normal scatter of results around the target), linearity (change in accuracy and precision with increased concentrations), range (concentrations of analyte which can be accurately determined), matrix effects (altered sensitivity of the method to the analyte due to the presence of other sample components), limit of detection (smallest concentration or amount of the analyte that can be detected), and limit of quantification (minimum analyte concentration or amount that can be accurately quantified). Of these, precision, or the agreement of a set of results, is of particular importance for evaluating the performance of analytical methods or individuals. As precision describes agreement, it is inversely related to the standard deviation (SD). The term imprecision describes how an increase in SD impacts this agreement; imprecision increases as SD increases, while precision decreases. Precision measurements can be classified as follows: repeatability is the variation in results generated by the same analyst running the same sample within a short time using the same method/equipment/material, and should have the lowest variation; replicatability or within-lab reproducibility is the variation seen after changing at least one of these previously controlled variables, and reproducibility measures variation after altering many or all of these variables, and should be the most inconsistent situation with the maximum variation in precision (Butzke and Ebeler 1999, Garfield et al. 2000, Horwitz and Albert 2006). Reproducibility and across-laboratory precision are interchangeable terms (Garfield et al. 2000). A useful and common expression of precision is the coefficient of variation (CV), which is the SD divided by the average value, or the relative standard deviation (RSD), which is the CV expressed as a percentage (Garfield et al. 2000). Additionally, there is a concept of analytical “ruggedness” or “robustness,” which is the ability of a method to tolerate common variations in technique, materials, or operating conditions both within and among laboratories, and still deliver good precision (Garfield et al. 2000).
Collaborative laboratory data in the wine industry
Early collaborative wine analysis studies were primarily done in conjunction with the Association of Official Analytical Chemists (AOAC) (Caputi and Wright 1969, Vahl and Converse 1980) or by researchers evaluating specific analytes. These studies were short-term programs aimed at validating new analytical methods prior to industry acceptance or adoption by the AOAC as Official Methods. Two multi-analyte collaborative proficiency studies were conducted in 1965 and 1975; relevant results include RSDs for alcohol (1.2 and 5.1% in 1965 and 1975, respectively), total (titratable) acid (1.9 and 7.0%), volatile acid (18.2 and 29.2%), total SO2 (16.3 and 12.2%), free SO2 (45.8 and 20.1%), and reducing sugar (4.5 and 19.6%). pH performance was expressed as the SD (0.2 and 0.1 pH units, Wildenradt and Caputi 1977). Although improvements were observed for some analytes due to implementation of new technology, the authors bemoaned the deteriorating performance for many analytes over the 10 years between studies, which they attributed to sloppy analytical technique.
The Horwitz ratio (HorRat) for evaluating analytical performance
Beyond providing insight into current analytical performance, data from proficiency testing can also indicate potential room for improvement in across-laboratory reproducibility (AOAC 2012). The reproducibility (expressed as RSD) for a given analyte increases with decreasing analyte concentration; this relationship can be empirically described by the Horwitz equation. If the concentration (C) of the analyte is expressed as a dimensionless mass fraction, with aqueous solutions substituting for the acceptable concentration factor of g/mL, then the empirically predicted RSD is equal to 2 × C −0.1505 (Horwitz and Albert 2006). The HorRat is the ratio of the observed RSD to the predicted RSD. The HorRat has been adopted by the AOAC, is in use by agencies such as the U.S. Department of Agriculture, and is being evaluated in the European Union. Generally, HorRat values between 0.5 and 2.0 indicate satisfactory industry-wide performance, but the decision on which value to accept can be influenced by regulations, industry standards, financial or legal risks, or analytical costs needed to improve the ratio (Horwitz and Albert 2006).
History and description of the Collaborative Testing Services (CTS) program
The CTS program has evolved over the years in response to technical and consumer feedback, but the overall concept has remained constant: two different wines of similar analytical composition are sent to subscribing laboratories and each laboratory analyzes the two wines using the procedures in use at that facility. Each laboratory reports their results within a specified time in duplicate, to capture within-laboratory variation. Coded results are returned to laboratories, which can identify their own results, but the data from other laboratories remains anonymous. Reports contain comparative performance values (CPVs), which are z-scores of individual laboratory results compared to the overall mean (“grand mean”) of all non-outlier results. For each analyte, results from the two wines are graphed on a two-sample Youden plot, which allows laboratories to readily evaluate systematic and random errors for each analytical measure. Tracking that error over multiple cycles can facilitate identification of sources of analytical errors. Data are classified as outliers and excluded from statistics when they are >3 SDs (σ) from the grand mean for a given sample. Data are flagged as warnings when they are >2σ from the grand mean for a sample or if a laboratory has exceeded what other laboratories have determined is an acceptable difference in values for the two samples. After a delay of some months, the coded results from each round are accessible to the public on the CTS website (www.collaborativetesting.com). As of 2014, routine testing includes alcohol, titratable acidity, pH, specific gravity, volatile acidity, free SO2, total SO2, residual sugar, glucose plus fructose, malic acid, absorbance at 420 and 520 nm, and copper.
Changes in the CTS program since its beginning in 1999 include reducing the number of annual cycles from four to three in 2001, reclassifying the residual sugar measurement into two separate tests based on significant differences in target analyte (residual sugar and glucose plus fructose) in Cycle 19 in 2005, adopting recommended units (Burns and Caputi 2002) in Cycle 22 in 2006, establishing a pattern of red, white, and blush wines as matrices in Cycle 23 in 2006, and additions to the standard panel of tests: absorbance at 420 and 520 nm in 2009 and copper in 2011.
This publicly available CTS data provides a good opportunity to evaluate industry and methodological performance, but only a few testing cycles have been evaluated. A thorough review of approaches to laboratory quality and an introduction to the first cycle of results (Butzke and Ebeler 1999) also set a high analytical performance goal, stating that commercial wine production should target a reproducibility (across-laboratory RSD) of 1% following outlier removal, as described above. These authors note that only pH and alcohol methods approached this criterion. Little improvement was noted after the first six cycles of the program (Butzke 2002), with only alcohol, specific gravity, and titratable acidity measurements deemed adequately reproducible. Poor reproducibility was noted for residual sugar, volatile acidity, malic acid, pH, and free and total SO2 analyses. The author also noted the impact of the combined errors in pH and free SO2 on the calculated value for molecular SO2. Unlike earlier researchers, no explanations for poor reproducibility were suggested, although improved performance in titratable acidity over earlier collaborative testing was attributed to increased use of autotitrators.
In this report, we review the first 13 years of data from the CTS program. As with earlier reports on collaborative testing, we present the overall performance for specific analytes and the bias and reproducibility of individual methods. By normalizing individual analytical results for each individual wine sample, we also compare results across all wines (and thus, all years), providing a more complete picture of ongoing performance over more than a decade despite individual wine sample chemistries. We also evaluate potential space for analytical improvements based on HorRat ratios. Finally, the large data set and extended length of the study allows us to evaluate the relative impact of method selection on analytical performance.
Materials and Methods
Raw data were entered into Microsoft Excel for 12 analytical tests (alcohol, titratable acidity, pH, free SO2, total SO2, malic acid, volatile acidity, residual sugar (combined methods), residual sugar (post-method separation), glucose plus fructose, specific gravity, and absorbance at 420 nm and 520 nm) for both wines from the CTS program in Cycles 2 to 40 (Spring 1999 to Spring 2012). When the CTS raw data included a laboratory-specified method or instrument information, it was preserved for each retained data point, as was wine color (red, white, or blush) and grape variety. Information obtained from the industry supplier of the wines included data on added sorbate in some wine samples, primarily blush wines. These data (red, white, blush, and sorbate-positive) were classified as dummy variables.
Laboratories were asked to report results in duplicate. Averages of the two values are considered one data point, and the SD of the two replicates was preserved for later within-lab d (repeatability) calculations, unless the original data was deemed an outlier, in which case it was removed.
Data were expressed in recommended standard units (Burns and Caputi 2002) and data from cycles prior to 2006 were converted to these standard units when necessary. In the 2005 cycle, residual sugar was divided into residual sugar and glucose plus fructose, and the glucose plus fructose data were analyzed separately. pH results were converted to molar activities of hydrogen ions and analyzed both as pH and as hydrogen ion activity, as logarithmic values are not well suited to statistical operations.
Outliers were removed by repeated application of the four-sigma rule. Within each cycle and for each wine, mean values and SDs (σ) for each analyte (across all methods) were calculated and results that differed from the mean by more than four sigma (|observed − mean| >4σ) were removed. This process was iterated until no values were >4σ from the mean. This criterion is less stringent than the three-sigma criterion used by CTS for outlier detection because we wanted to better characterize existing analytical variation. The primary effect of outlier removal in our current study was to eliminate gross errors arising outside of the analytical method, such as those due to errors in data entry, unit conversion, “powers of ten,” or related transcription issues.
Following outlier removal, descriptive statistics: mean, SD (within-laboratory and across-laboratory), and relative SD were calculated for each analyte of the 78 individual wine samples. Mean values for each data point from each individual wine were used to calculate z-scores for all individual analyses of that wine, rather than using a grand mean across all 78 wines. The resulting individual wine-weighted analyte z scores were compared across all wine samples to give the complete distribution of the targeted analyte results across all wines used in the 39 testing cycles for a maximum n of 78. The purpose of this approach was to find the variation in the imprecision of the analyses, rather than to find the variation in wine composition. These data reflect the overall industry performance for each analyte regardless of method or instrument used.
Histograms and linear regression of these individual-wine z-score values of analyte means and their corresponding within- and across-laboratory SDs were made to compare imprecision. Additionally, both within-laboratory and across-laboratory SDs and RSDs were compared to the analyte concentration to provide information about how concentration affects imprecision for each analyte.
Concentration factors for the Horwitz equation were calculated using mean analyte concentration values for each individual wine and HorRats were determined for titratable acidity, alcohol, free and total SO2, volatile acidity, malic acid, and the sugar measurements: residual sugar prior to 2005, when all sugar methods were not distinguished, glucose plus fructose after methods were split in 2005, and residual sugar after methods were split in 2005.
To characterize the wine sample matrices, one-way ANOVA was performed with dummy variables (red, white, and blush) against analyte mean values. To represent the covariance of the analytes and the matrices, principle component analysis (PCA) of the analyte z-scores and a dummy variable of sorbate addition was performed. To evaluate if wine matrix or composition contributed to imprecision, Pearson’s correlations were run using average analyte values and dummy variables (red, white, blush, and sorbate-positive) against the normalized across-laboratory SDs. Absorbance at 420 and 520 nm were excluded from the correlations due to their limited data set.
Finally, to determine whether method or instrumentation choices affected reproducibility, individual z-scores for every data point, along with self-reported information on method and/or instrumentation, were generated. These data also allowed evaluation of changes in method or instrumentation over the course of the study. Histograms, linear regression, and ANOVA were made on these individual-analysis data. When needed, the z-scores were converted back to analyte concentration equivalents using the appropriate sample mean and SDs.
Minitab statistical software (Minitab 16.2.4; Minitab Inc., State College, PA) was used for chi square analysis, regression analysis, correlations, ANOVA, PCA, Tukey’s significant difference, and for plotting histograms.
Results and Discussion
Participation
Participation in the CTS wine analysis proficiency testing program has ranged from 30 to 77 participants per cycle, with an average of 55 contributing results per cycle. During the first four years of the program, the number of reporting laboratories ranged from 30 to 60; during the past four years, this figure has grown to 60 to 77. The identities of the participating winery laboratories are secret and protected, but it can be assumed that there are three subsets of participating laboratories: those with formally accredited quality control systems (or those with at least sound laboratory quality systems concepts in place), those with few or no laboratory quality systems in place (notwithstanding experienced personnel), and those with neither training, experience, or knowledge of laboratory quality systems. Anecdotal observations by administrators of the program suggest that some of the latter are occasional “visitors” to the program who participate randomly; this is based on the higher number of outliers submitted by infrequent participants than by ongoing participants.
Outlier removal and distribution of remaining data
As expected, the repeated four-sigma outlier removal method identified fewer outliers (2.9%) than the three-sigma method used by CTS (10.8%). Malic acid, glucose plus fructose, and specific gravity had relatively more outliers, while pH and free SO2 had fewer (χ2 test, p < 0.05). No significant correlation was observed between analytical method or instrumentation and likelihood of outlier removal for any parameter (χ2 test, p < 0.05). Outliers largely appeared to be due to clerical, mathematical, or unit conversion errors, e.g., a factor of 10 differences between mean and outlier, rather than due to method-specific issues.
Following outlier removal, histograms of the individual-wine weighted z-scores for each analyte typically appeared unimodal, although most distributions were leptokurtic and many showed left or right skews (Figure 1). The higher kurtosis is expected from distributions made using four-sigma data selection. Most striking was a deviation from the unimodal pattern that suggested some potential problems with analytical nomenclature. The bimodal distribution for specific gravity (Figure 1G) may arise from operator confusion between specific gravity and density; the location of the smaller mode is 0.0018 specific gravity units lower than the mode of the larger distribution, or approximately the error expected if density values were reported instead of specific gravity.

Frequency distribution of individual-wine weighted z-scores for the analysis parameters (A) alcohol; (B) titratable acid; (C) volatile acid; (D) total sulfur dioxide; (E) free sulfur dioxide; (F) malic acid; (G) specific gravity; (H) pH; (I) hydrogen ion activity; (J) residual sugar (all methods pre-split); (K) residual sugar (post-split); (L) glucose plus fructose; (M) absorbance at 520 nm; and (N) absorbance at 420 nm.
Other deviations from symmetrical distributions may arise from methodological biases where multiple methods are used. For example, the positive skew for residual sugar prior to the analytical separation of cycle 19 in 2005 (Figure 1J) may occur because the two primary analytical methods, copper reduction assays and enzymatic assays, have different selectivities, with the former measuring other reducing compounds in addition to fructose and glucose (Amerine and Ough 1980, Iland et al. 2004). This would result in two overlapping distributions, with the enzymatic methods having a slightly lower mean. This issue resulted in separating assays for residual sugar (Figure 1K) and glucose plus fructose (Figure 1L) into different reporting categories after 2005 (hereafter referred to as pre- and post-split). The average residual sugar concentration was ~1 g/L higher than the glucose plus fructose concentrations in post-split measurements (Table 1). Both post-split categories demonstrate more symmetrical distributions than the earlier data, but residual sugar now shows a negative skew, indicating that this terminology is still used inconsistently among laboratories. Other positively skewed distributions, such as for titratable acidity, volatile acidity, and free SO2 (Figure 1B, C, E), likely arise from biases among methods and are discussed in more detail later.
Average wine analyte values, within- and across-laboratory standard deviation (SD) and relative standard deviation (RSD), and the ratio of the SD sources for 39 cycles of collaborative testing data.
Overall performance: SD and RSD
To estimate the overall performance over time, the individual wine values for means (μ) and SDs (σ) of the analytes were averaged across all wines and the reproducibility was expressed as a percentage (σ/μ), the across-laboratory RSD. Within-laboratory precision was calculated by CTS from duplicate analyses for each wine and also reported as SD and RSD (Table 1). Generally, reproducibility was comparable to results from collaborative studies of earlier decades: alcohol RSDs of 1.2 and 5.1% (earlier studies) versus 1.4% (current study); titratable RSDs of 1.9 and 7.0% versus 4.2%; volatile acid RSDs of 18.2 and 29.2% versus 17.5%; reducing sugar RSDs of 4.5 and 19.6% versus 18.3%; and pH SDs of 0.2 and 0.1 versus 0.042 for the 1965, 1975, and current studies, respectively (Wildenradt and Caputi 1977). CTS testing is open to any interested laboratory, while these earlier collaborative studies used specific, pre-selected participants. One possible explanation for the apparent lack of improvement in reproducibility could be the less restricted approach to winery laboratory inclusion in the CTS data.
The across-laboratory reproducibility can be compared with the within-laboratory repeatability to show the relative impact of typical sources of imprecision. The across-laboratory values were consistently higher, as expected, ranging from approximately three to eight times for most analytes to greater than 10 times the within-laboratory variation (Table 1). The very high relative variations in sugar, malic acid, and absorbance at 420 and 520 nm results, all of which are primarily (but not exclusively) tested using spectrophotometric methods, open the possibility that spectrophotometer calibration might be responsible for some across-laboratory performance issues; laboratory-to-laboratory variation in these instrument calibrations could explain the lower within-laboratory error and the very high across-laboratory error. Other instrument-based errors related to these analytes could include issues with pipet calibration, selection or availability of different cell path lengths, or calibration data set of infrared spectrophotometers. Method-based errors involving incomplete or slow enzymatic reactions, or impact of dilutions, may also be a factor. Calibration of equipment, use of standards and blanks, and testing the method using different sample sizes or concentrations are all ways to locate such systematic errors (Skoog et al. 1992).
HorRats applied to wine collaborative data
HorRats represent the ratio of observed to empirically predicted precision and may be calculated from the analyte concentration. Mean and SD HorRat values were calculated across all individual wine samples for the various analyses (Table 2). Typical targets for mean HorRat values recommended by international analytical organizations range from 0.5 to 2.0. In our study, only three parameters achieved this criterion when averaged across all samples: alcohol, titratable acidity, and total SO2. All other analytes that measure concentrations had overall average HorRat values above 2.0. It is not possible to calculate the Horwitz values for pH, specific gravity, or absorbance because these values are not concentrations. Individual HorRat values for each individual wine over the 39 cycles show that HorRat values are not consistent: even most analytes with average HorRat values below 2.0 have occasional samples above this value, represented by the percentage of times the HorRat value is above 2.0 for each analyte (Table 2). Only titratable acidity had a HorRat value consistently below 2.0 over 13 years; other analytes have HorRat values above 2.0 over 79% of the time (volatile acid, malic acid, and all analytical methods for sugar). Actual HorRat values for analyses of particular economic or regulatory importance can be well below the maximum recommended level of 2.0: inter-laboratory testing of milk fat and solids in the dairy industry has resulted in HorRat values of 0.1 to 0.4, possibly reflecting the great economic importance of these analytes (Horwitz and Albert 2006). For the wine industry, the analyte with the greatest economic impact may be alcohol, due to the critical maximum value of 14.04% for the lower “table wine” tax class of $1.07/gal (Alcohol, Tobacco Products, and Firearms) versus $1.57/gal if between 14.05% and 21% alcohol (Internal Revenue Code of 1986b). Although alcohol is one of the three analytes with an average HorRat across all 78 wine samples below 2.0, it nevertheless had 10% of wine samples with HorRat values above the maximum recommended acceptable HorRat value. Total SO2, another regulated analyte, had only 8% of the 78 wines with HorRat values under 2.0. Volatile acidity, although also regulated, had an average HorRat of 3.0 and 79% of the individual wine samples with values above this. The highest HorRat values were found with malic acid and with all methods for analyzing sugar; although these are not regulated components, they have significant impact on the taste and stability of wine and improvement would be recommended. To our knowledge, this is the first time that the HorRat concept has been applied to wine analysis and it yields interesting insights into the potential for further improvements.
Horwitz ratio (HorRat) values for wine collaborative testing from 1999 to 2013.
Analytical methods used
Wine laboratories self-reported the methods or instruments used beginning at the eighth cycle. The percentage of data points used for each technique demonstrates the range of methods and instruments used over the study (Table 3). Most of the methods, techniques, or instruments listed are mentioned or described in common texts (Amerine and Ough 1980, AOAC 2012, Iland et al. 2004, Jacobson 2006, OIV 2004, Zoecklein et al. 1994). Although the versions published in these texts are similar, they are rarely if ever identical; therefore, use of any of these self-reported method descriptors does not imply that a specific protocol or instrument model was used.
Percentage of self-reported methods or instruments used for each analyte.
Wine matrix composition
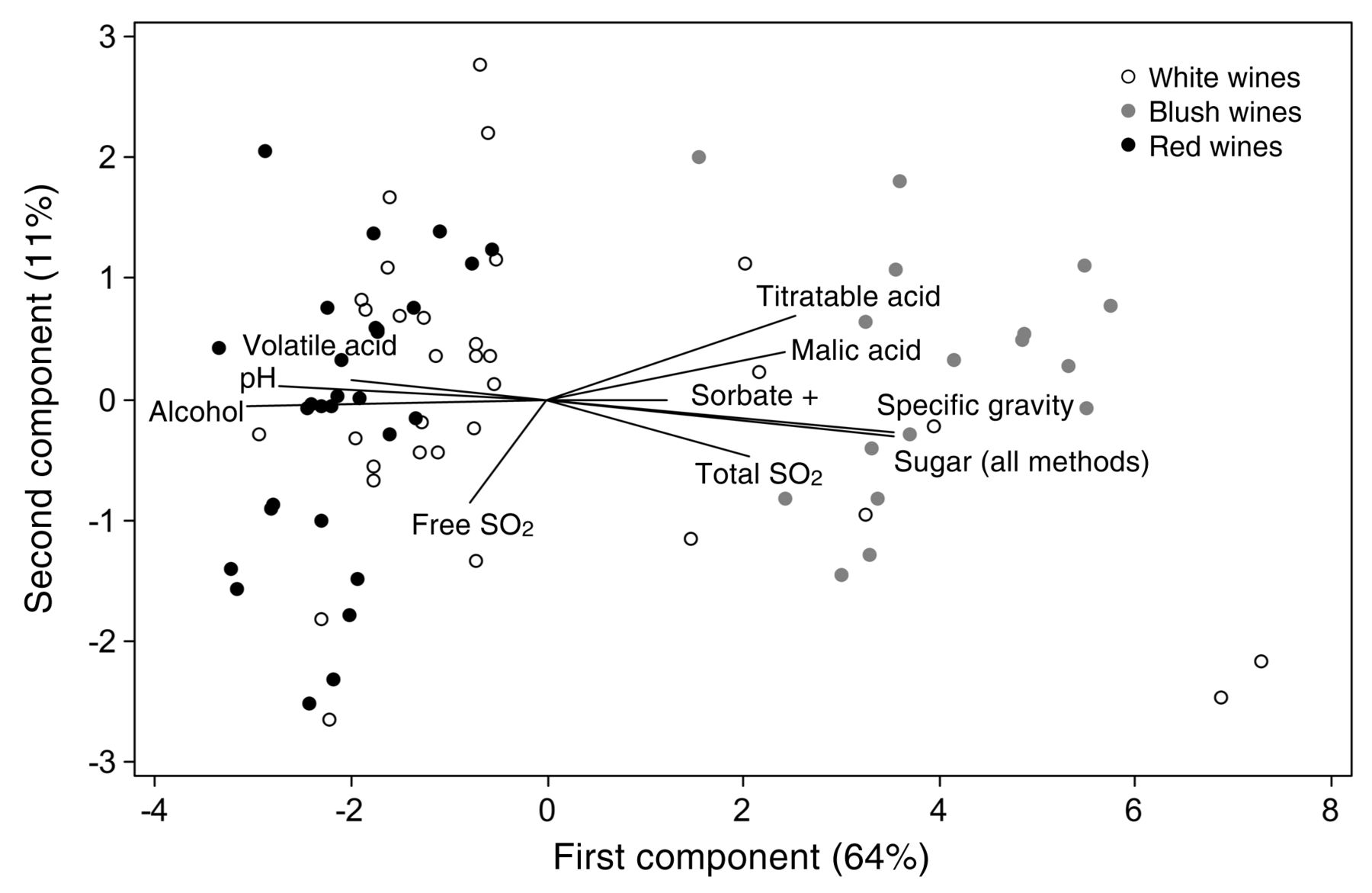
To characterize the typical composition of the sample matrix (red, white, or blush) for the wines used over the course of the study, the three matrices were classed as dummy variables and analyzed by one-way ANOVA for each analyte and for the known addition of sorbate (as a dummy variable). Tukey’s test demonstrated statistically significant differences for each analyte and provided a view of matrix typicity for this set of wines (Table 4). Red wines had significantly higher (and blush wines significantly lower) pH and concentrations of volatile acid and free SO2, while red and white wines had significantly more alcohol than blush wines. Red wines were significantly higher in absorbance at 420 and 520 nm than white or blush wines. Blush wines were significantly higher (and red wines significantly lower) in total SO2, malic acid, hydrogen ion activity, residual sugars (post-split), and glucose plus fructose. Blush wines were significantly higher than both red and white wines in titratable acid, specific gravity, and residual sugars (pre-split). These data characterize the composition of the wines used in this study and may or may not be typical of similar wines in the broader sense. PCA of the matrices using the z-scores of means of these analytes (excluding the absorbance values due to incomplete data and using a combined before/after split of sugar analyses to achieve coverage of all samples) plus a dummy variable of sorbate addition was also performed and indicated the covariance of many of the analytes, as expected (Figure 2). The primary separation was of red wines and blush wines on the first component, explaining 64% of the variation: red wines were associated with higher volatile acid, pH, and alcohol while blush wines were associated with higher titratable acid, malic acid, total SO2, specific gravity, sugar (by combined methods), and added sorbate. The second component explained 11% of the variation and was primarily associated with free SO2. Neither component could separate the white wines from the red and blush wines; presumably, this would occur if data on the absorbance at 420 and 520 nm data could be included, as color may be the primary analytical difference between white and non-white wines in this group of samples.
Wine matrix (red, white, or blush) average composition for the 78 wines tested. Tukey’s significant differences from one-way ANOVA of analyte mean values and dummy variables (p < 0.05).
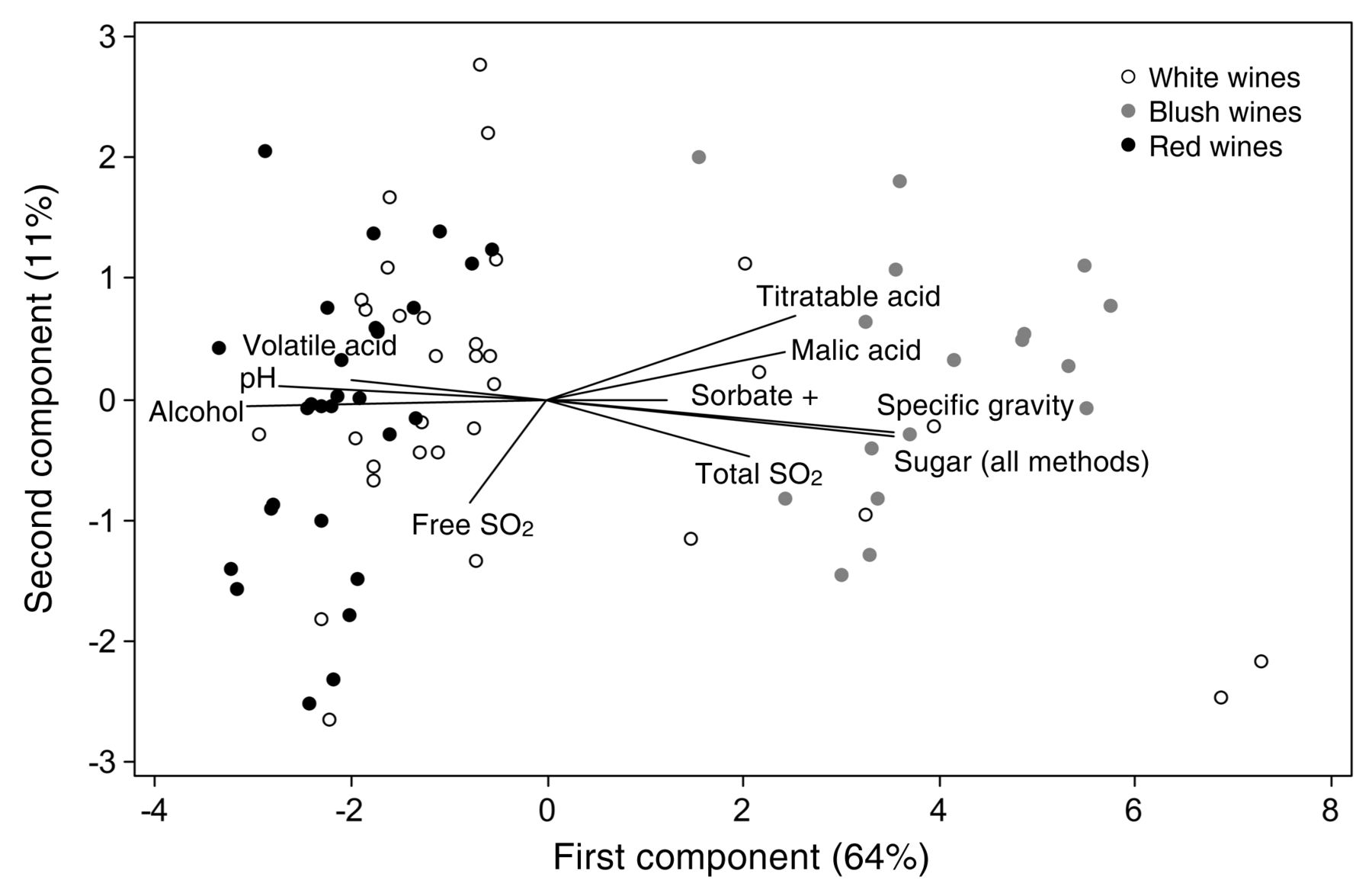
Principal components of the wine matrix using mean analyte composition for the 78 wines tested.
Wine matrix and analytical composition effects on imprecision
To evaluate the impact of wine matrix/composition on analytical performance, the dummy variables of wine matrix (red, white, and blush) along with the z-scores of the analyte composition were compared to the across-laboratory imprecision using Pearson’s correlation (Table 5). The matrices of red and blush wines had significant impact on precision for malic acid, hydrogen ion activity, and all measures of sugar, with red wines decreasing (improving) the imprecision for these analytes while blush wines increased (worsened) industry performance. The blush wine matrix increased (worsened) imprecision of volatile acid and titratable acid measurements. Notably, the white wine matrix had no impact on analytical imprecision. Individual analytical components also correlated significantly with reproducibility for all analytes, with the notable exception of alcohol. As alcohol was the analyte with the best performance characteristics, this is not unexpected. Several analytes (titratable acid, free SO2, total SO2, malic acid, hydrogen ion activity, and all sugar analyses) showed concentration effects and will be discussed separately. Specific method bias will also be discussed later.
Pearson correlation of wine matrix and composition effects on across-laboratory imprecision (inclusive of all methods) for the individual wines. Coefficients shown are significant at p < 0.05. A420 and A520 excluded due to smaller data set. N = 78 for all except residual sugar pre-split (n = 34), residual sugar post-split (n = 44), and glucose plus fructose (n = 44).
In some instances, the correlation between matrix composition and precision for an analyte may indicate a causative effect, although in others, the correlation may be due to covariance. For example, across-laboratory imprecision of volatile acidity analysis increased with increasing specific gravity, malic acid, total SO2, and titratable acid. Volatile acidity imprecision also increased in blush wines and those with added sorbate; imprecision decreased as alcohol increased and there was no significant impact of volatile acid concentration. Of these, sorbate and total SO2 are well known to interfere with determining volatile acidity using a Cash steam still method (Zoecklein et al. 1994). This factor is discussed more in the section on methodological bias. Because the IR spectra of acids and sugars are similar, volatile acid analysis using IR methods can be affected by any other acid and by sugars, especially if the target analyte concentration is approaching the IR limit of detection of 0.2 g/L (Bauer et al. 2008).
Titratable acidity imprecision increased with sugar levels (using any parameter), specific gravity, total SO2, malic acid, and hydrogen ion activity. Titratable acid imprecision also increased in blush wines and those with added sorbate; imprecision decreased with volatile acidity, alcohol, free SO2, and pH. Known interferences with the titratable acid methods include the endpoint determination and the presence of carbon dioxide for titration-based methods (Guymon 1963). While it is possible that these parameters are interferences that somehow increase or decrease imprecision, it appears more likely that these parameters co-vary with titratable acid (Figure 2) and that error in titratable acidity measurements by titration increases with concentration. IR-based methods for titratable acid have similar issues, as samples must be degassed, and both sugar and other acids can interfere with the measurements (Bauer et al. 2008).
Industry imprecision for free SO2 increased with total SO2 and with free SO2, showing an effect of concentration. Total SO2 imprecision increased with total SO2 and also with volatile acidity. Volatile acidity is a known potential interference when determining total SO2 with the aeration oxidation method (Rankine and Pocock 1970). Malic acid industry imprecision increased in blush wines and with sorbate additions, and with increasing titratable acid, total SO2, specific gravity, all measures of sugar, hydrogen ion activity, and malic acid (showing a concentration effect). These factors are typical of wines which do not undergo malic conversion as discussed earlier in reference to the matrix (Table 4); furthermore, malic acid imprecision decreased with increasing pH, free SO2, alcohol, volatile acid, and in the red wine matrix. As all these are typical characteristics of wines that undergo a malolactic conversion, their correlation with malic acid imprecision is more likely due to covariance rather than a direct causative effect.
The industry imprecision in pH and the related hydrogen ion activity is interesting, as the only significant correlation with increasing pH imprecision is with a decrease in volatile acid. Yet, hydrogen ion activity imprecision increased with increasing titratable acidity, total SO2, malic acid, specific gravity and the measures of sugar, in addition to increasing hydrogen ion activity (a concentration effect). Hydrogen ion activity imprecision also increased in blush wines and with added sorbate; the imprecision decreased in red wines and with increasing volatile acidity and alcohol (and pH). These correlations suggest that hydrogen ion measurement precision is concentration-dependent and that the analyses performed by the industry are less precise at lower pH values (i.e., higher hydrogen ion activities). This result is discussed further below. The imprecision experienced by the industry for the specific gravity measurements increased in wines with sorbate added and decreased in wines with higher volatile acid, possibly indicating that the sweeter blush wines introduced more imprecision than did red wines.
Finally, all industry measurements of sugar showed the same correlations with imprecision: all sugar imprecision increased with increasing titratable acid, total SO2, hydrogen ion activity, and specific gravity; all showed increasing imprecision with increasing concentration of sugar (a concentration effect), and all had imprecision increase with blush wines and with sorbate additions. All sugar measurements saw decreases (improvements) in imprecision with increases in pH, free SO2, volatile acidity, and in the red wine matrix. These factors may indicate that concentration is a primary factor and these other parameters are covariates with the sweeter blush wines.
Performance over time
Changes in the analytical performance of the subscribing labs show a variety of historical trends. Over the 13 years of data, alcohol analysis results have significantly (p < 0.01) improved in precision, with an average decrease in SD of 0.0028% v/v per cycle (Figure 3); alcohol RSDs also showed significant improvement. This increased precision in alcohol analysis may be due to the adoption of IR-based methods for alcohol measurement. In contrast with the improved precision of alcohol analysis, significant (p < 0.01) loss in performance (increased imprecision) was found for both titratable acidity and pH over the 39 cycles, with an average increase in SD of 0.0027 g/L tartaric acid or 0.0003 pH units per cycle, respectively. Malic acid imprecision (as SD) also increased significantly (p < 0.05), at a rate of 0.0044 g/L malic per cycle, yet the RSD for malic did not change significantly over the same period, indicating that the loss in precision may be a proportional error. This loss in performance for titratable acidity, pH, and malic acid is surprising, as increases in technology (specifically, the increase in the use of autotitrators) were previously assigned responsibility for increased precision (Butzke 2002). No other significant improvements or losses in performance were found. However, the variation in reproducibility for the same parameter across multiple cycles was striking. For example, reproducibility for titratable acidity varied from 0.15 to 0.5 g/L across all cycles (RSD range, 2 to 7%). Similar effects were observed in other parameters (Supplementary Material). In some cases, cycle-to-cycle variation may arise from wine matrix effects as discussed earlier, but could also be due to variability in participating wineries. Regardless, this observation cautions against using the results of a single testing cycle to draw conclusions about methodological or laboratory performance.

Across-laboratory precision (reproducibility) over 39 cycles for (A) alcohol standard deviation (SD, % v/v); (B) titratable acid SD (g/L); (C) pH SD; (D) malic acid SD (g/L); (E) alcohol relative standard deviation (RSD = SD/concentration); (F) titratable acid RSD; (G) pH RSD; and (H) malic acid RSD. Alcohol SD = 0.22 − 0.0028×, R2 = 0.44 (p < 0.01); alcohol RSD = 0.018 − 0.0002×, R2 = 0.20 (p < 0.01); titratable acidity SD = 0.20 + 0.0027×, R2 = 0.1642 (p < 0.01); titratable acidity RSD = 0.03 + 0.0004×, R2 = 0.1772 (p < 0.01); pH SD = 0.037 + 0.0003×, R2 = 0.13 (p < 0.01); pH RSD = 0.01 + 0.0001×, R2 = 0.179 (p < 0.01); malic acid SD = 0.12 + 0.0044×, R2 = 0.074 (p < 0.05); malic acid RSD = 0.19 + 0.003×, R2 = 0.03 (ns). ns = not significant.
Concentration dependence of imprecision
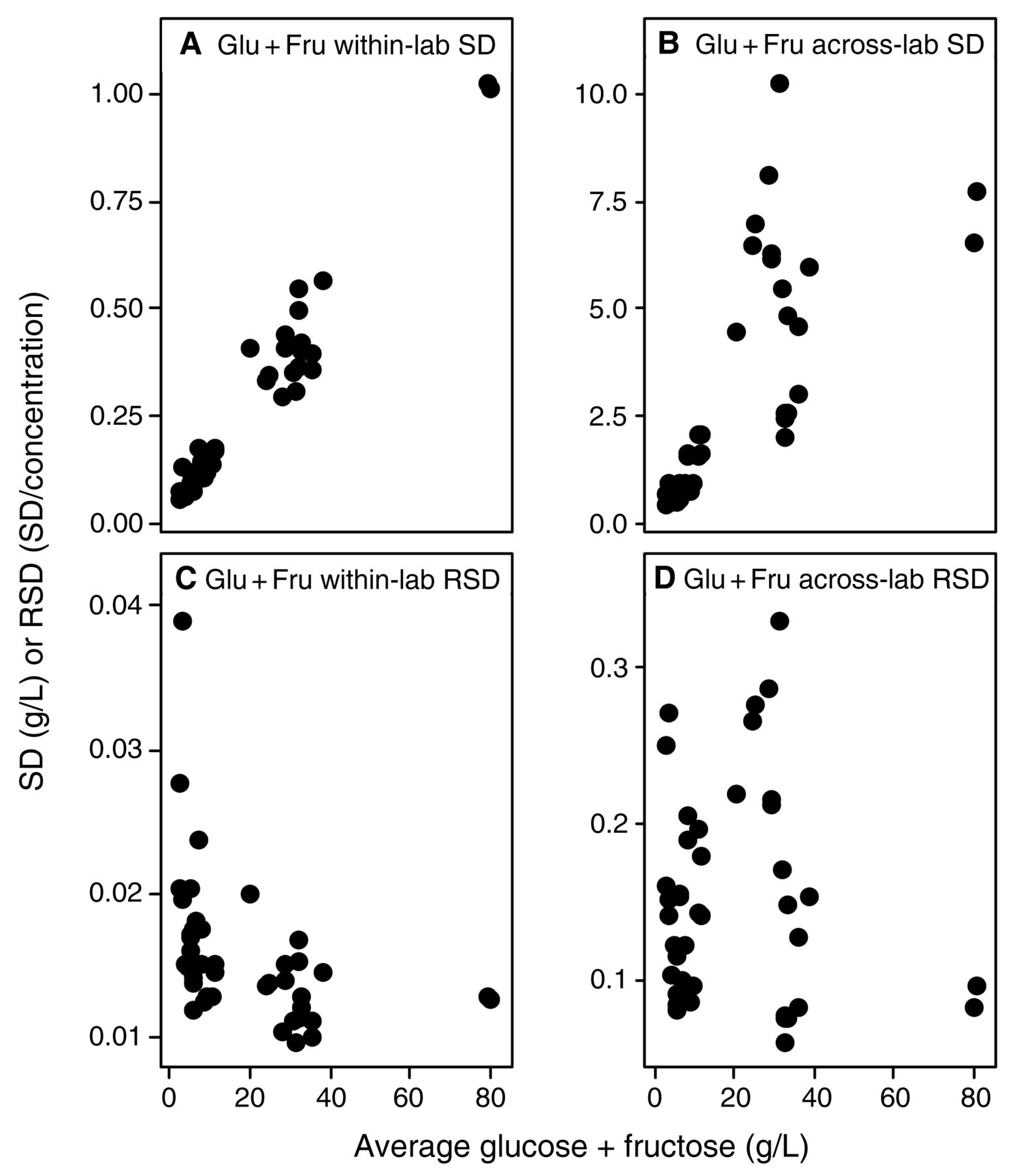
Evaluating the across-laboratory imprecision against analyte concentration provides information about relative concentration dependence of the errors; that is, whether they are constant or proportional errors, which may indicate how to find and control the sources of variation. For example, systemic errors such as those introduced from titration endpoints have an error which is constant, but have a relative error which varies when sample size is changed. In these examples, the SD/concentration curve would be constant with increasing concentration, but the RSD/concentration curve would reflect this constant error by decreasing with increasing concentration; thus, these constant errors can become problematic at very low concentrations. Similarly, interfering substances in IR measurements would behave as a constant, compounded at lower target analyte concentrations that approach the lower limit of the methods (Bauer et al. 2008). If the imprecision is a proportional error, then errors introduced by multipliers are likely sources of the error (Skoog et al. 1992), e.g., dilution steps, volume measurements, enzymatic reaction times, or interfering contaminants. For such proportional errors, relative error (RSD) remains constant with increasing concentration, while the absolute error (SD) increases with concentration. Alternatively, upper and lower limits of the methods used may have been exceeded without operator knowledge. Within- and across-laboratory SDs and RSDs for each wine were plotted against the analyte mean values to determine if measurement imprecision expressed as SD or RSD (absolute or relative error) was concentration-dependent. Summary data for slope and correlation coefficients of within-laboratory (repeatability) (Table 6) and across-laboratory (reproducibility) (Table 7) versus concentration are provided. Several parameters had errors that correlated with concentration (p < 0.05 and R2 > 0.7); these correlations were strongest for malic acid and glucose plus fructose (Figures 4 and 5). Both analytes show increasing SD with increasing concentration, indicating a proportional error. However, the sharp increase in RSD versus concentration at low concentrations indicates the presence of a constant, low-level source of error in malic acid analyses at <0.5 g/L (Figure 4) and to a lesser extent with glucose plus fructose (Figure 5). The concentration-dependent error is most easily explained by the dilution steps typically necessary for enzymatic/spectrophotometric analysis of malic acid and glucose plus fructose, or, alternatively, by interferences of acids and sugars in IR methods, along with loss of precision as the malic acid concentration approaches the lower limits of detection for this method. The poor reproducibility (average RSD = 35%, max 100%) for malic acid at concentrations <0.5 g/L may reflect the noise limit of typical methods, and is problematic since malic acid measurements at these concentrations are often necessary to evaluate whether malolactic fermentation is complete (Butzke 2010). This reduced performance at low malic acid concentrations indicates a need among wineries to review protocols used in the commonly employed enzymatic analysis method (dilution protocols, sample sizes, enzyme concentrations and reaction times, and instrument calibrations), or to be aware when IR methods are at lower limits. These issues were less apparent for glucose plus fructose analyses because no wines under study had concentrations <2.5 g/L (as compared to minimum malic acid concentrations of <0.1 g/L). In contrast, SDs for many other parameters such as volatile acidity, titratable acidity, specific gravity, alcohol, and total and free SO2 were either weakly or not correlated with concentration. In some instances, the results had negative correlations of concentration with RSD, indicative of constant sources of error (Tables 5 and 6). Notably, analytes that display concentration-independent errors were typically analyzed by methods that do not require sample dilution steps and the critical source of error may be either constant interferences, challenges in defining the endpoint, or other issues typical of a consistent systematic error. Finally, pH measurement, when evaluated as hydrogen ion activity, shows a concentration-dependent error. Speculatively, this may arise from wineries calibrating their pH meters with pH 4 and 7 solutions as typically recommended in wine texts (Iland et al. 2004, Zoecklein et al. 1994) rather than over the typical pH range of wine (3 to 4); this would provide an interesting avenue for recommended improvements to the method in future work.
Concentration dependence of within-laboratory (repeatability) precision. Regression of standard deviation (SD) and relative standard deviation (RSD) against average analyte concentration for individual wines.
Concentration dependence of across-laboratory (reproducibility) precision. Regression of standard deviation (SD) and relative standard deviation (RSD) against average analyte concentration for individual wines.
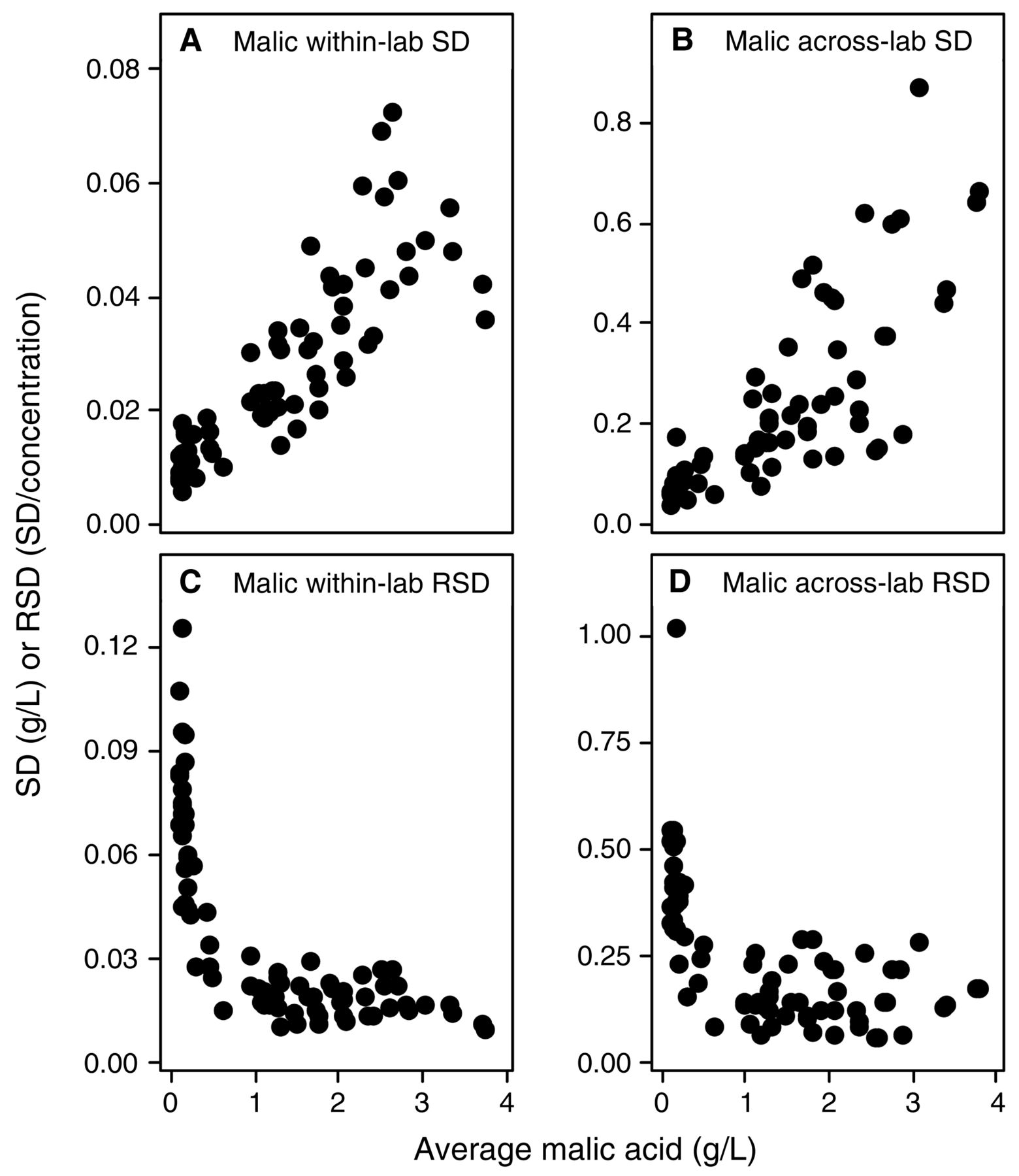
Within-lab and across-lab standard deviation (SD) and relative standard deviation (RSD) for malic acid concentration (n = 78). (A) within-lab SD; (B) across-lab SD; (C) within-lab RSD; and (D) across-lab RSD. SD units are in g/L; RSD is unitless (SD/concentration).
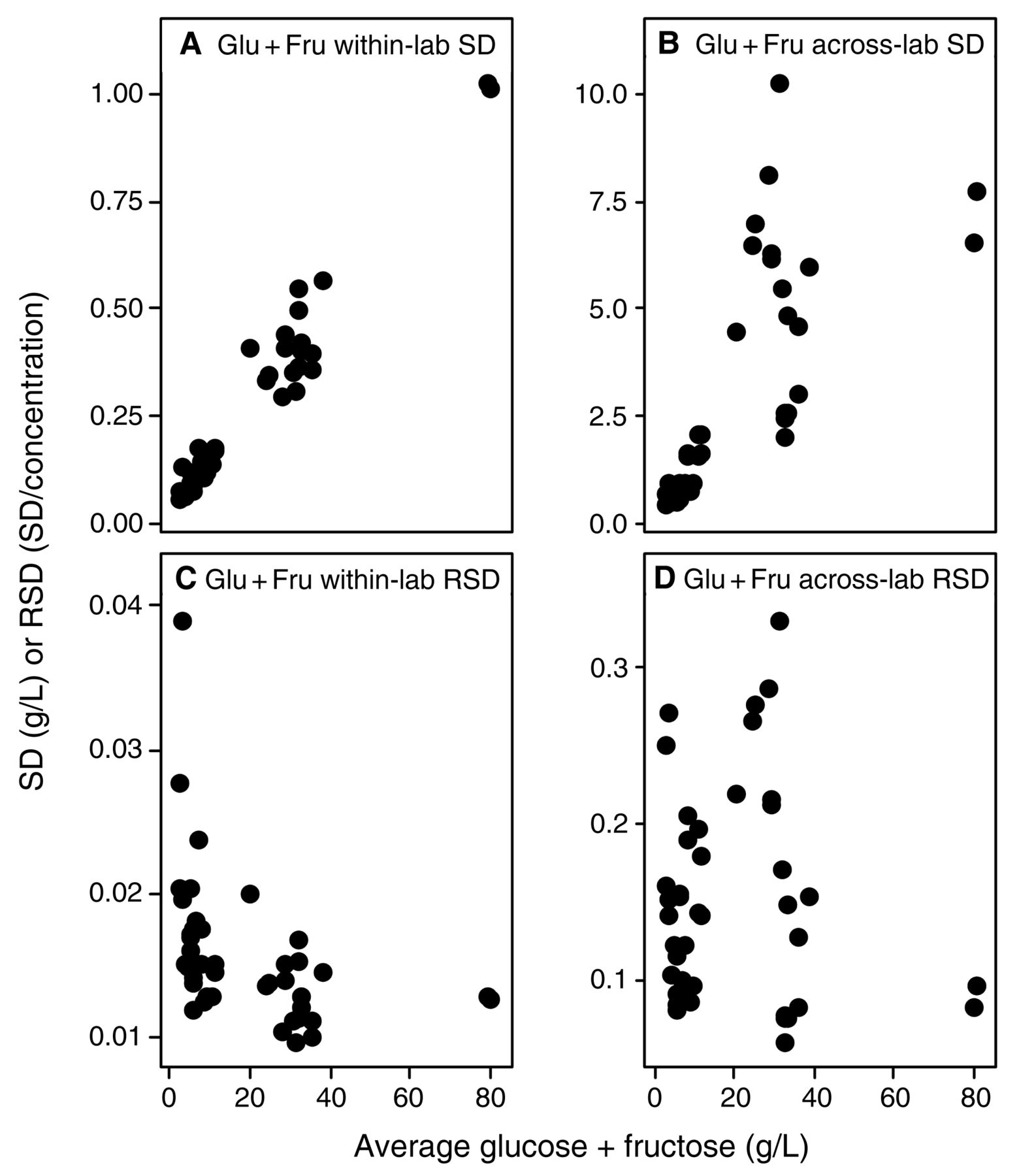
Within-lab and across-lab standard deviation (SD) and relative standard deviation (RSD) for glucose plus fructose concentration (n = 42). (A) within-lab SD; (B) across-lab SD; (C) within-lab RSD; and (D) across-lab RSD. SD units are in g/L; RSD is unitless (SD/concentration).
Changes in methods used over time
Several analytical parameters were analyzed using multiple methods across wineries (Table 3). To evaluate whether the method use frequency changed over time, the percent usage of each method used more than 2% of the time was plotted against cycle and trend analysis parameters calculated using least squares regression (Table 8). Method self-reporting did not begin until Cycle 8. Five analytes showed significant changes in method/instruments over the course of the study: alcohol, titratable acid, volatile acid, free SO2, and total SO2. Use of IR equipment (near infrared [NIR] and Fourier transform infrared [FTIR]) and distillation/density for alcohol analyses increased by 0.73, 0.54, and 0.42% per cycle, respectively, at the expense of ebulliometry and gas chromatography (−0.91 and −0.73% per cycle). Use of the Cash still to determine volatile acidity declined by 0.95% per cycle, as use of enzymatic methods (for acetic acid), segmented flow, and FTIR increased by 0.26, 0.26, and 0.47% per cycle, respectively. Manual titrations for titratable acid declined −0.36% per cycle as FTIR use increased 0.35 % per cycle. Segmented flow, flow injection, and other colorimetric methods for free SO2 analysis increased by 0.12, 0.36, and 0.19% per cycle, respectively, at the expense of aeration oxidation (−0.42% per cycle). Use of segmented flow, flow injection, and other colorimetric methods for total SO2 analyses increased by 0.24, 0.31, and 0.31% per cycle, respectively, at the expense of Ripper (−1.0% per cycle). Despite the increase in automation and technology over the course of the study, no increases in precision were found except as noted for alcohol analysis.
Changes in self-reported method use over cycles 8 to 40, for methods with >2% average reported use. Regression analysis of percent reported use of method is reported by cycle number.
Relative method accuracy and method bias estimates
Although the CTS proficiency scheme can’t provide “true” values due to the nature of the samples, it is useful to provide some information on the accuracy of the methods used. Mean z-scores by method, SDs, and a conversion value for z-score into analyte unit value are provided (Table 9). The methods for absorbance at 420 and 520 nm were not analyzed because of the small sample size. Glucose plus fructose methods and residual sugar methods showed no significant differences among methods used (excluding the “other” or “unassigned” methods), which is in part a consequence of the poor reproducibility of the methods (HorRat >2 for >90% of samples) and also because of the diminished statistical power due to the splitting of the methods at cycle 19. One exception to this statement is that prior to the split of methods, the enzymatic method gave significantly lower (−0.35 g/L) residual sugar results than the HPLC, FTIR, and copper reduction methods.
Z-scores and standard deviations (SD) for self-reported methods for each analyte. Within an analyte category, shared letters within a Tukey grouping indicate that the methods were not significantly different (p < 0.05).
To evaluate whether differences existed among methods for a given analyte, distributions of analyte z-scores for each method were plotted as histograms showing the deviation from the overall (combined methods) analyte mean (Figures 6 to 12). While these results are interesting for discussion of differences among methods, we caution that they provide no certain information on which methods are the most accurate because the samples were not reference materials with known values. Because the method with the most analyses will dictate the mean value for a given analyte, that same method will usually dominate the data; proximity to the mean is thus not an indication of superior accuracy.
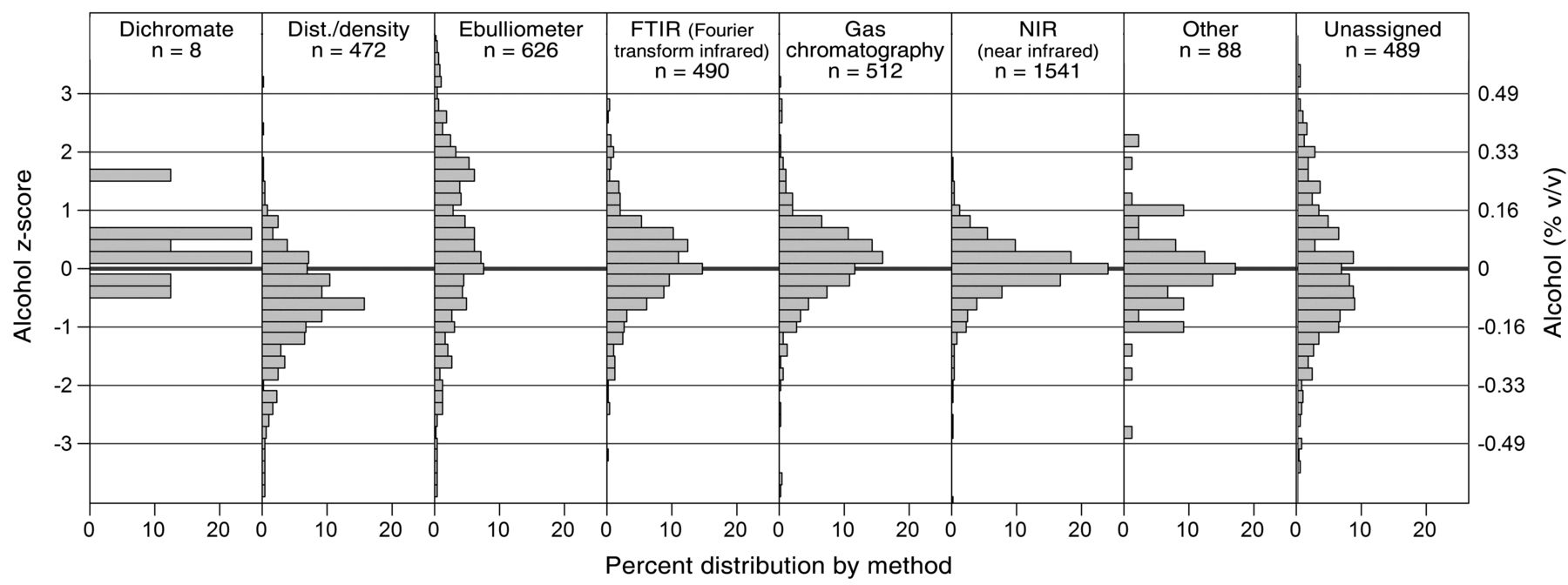
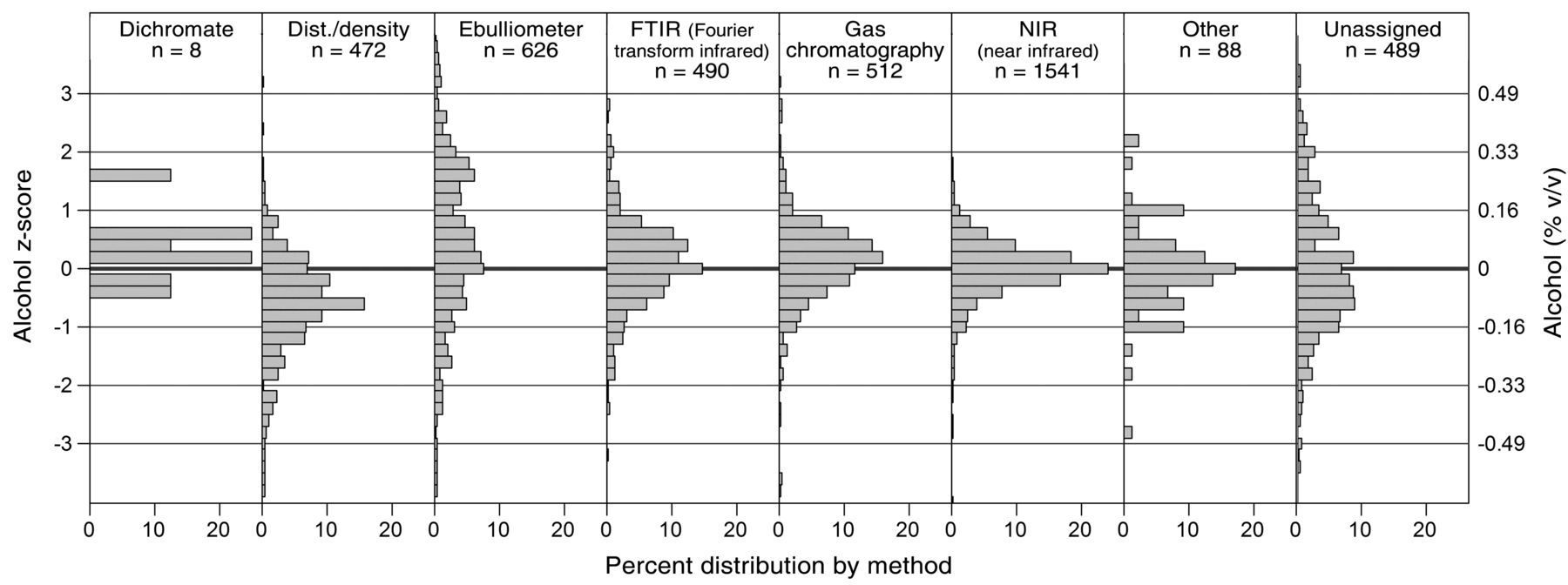
Z-score distributions of alcohol concentrations by self-reported alcohol analysis method. The left axis is the z-score and the right axis is equivalent units. Scores were normalized by individual wine results to allow analytical comparison across all wines.

Z-score distributions of total SO2 concentrations by self-reported total SO2 analysis method. The left axis is the z-score and the right axis is equivalent units. Scores were normalized by individual wine results to allow analytical comparison across all wines.

Z-score distributions of free SO2 concentrations by self-reported free SO2 analysis method. The left axis is the z-score and the right axis is equivalent units. Scores were normalized by individual wine results to allow analytical comparison across all wines.

Z-score distributions of volatile acid concentrations by self-reported volatile acid analysis method. The left axis is the z-score and the right axis is equivalent units. Scores were normalized by individual wine results to allow analytical comparison across all wines.

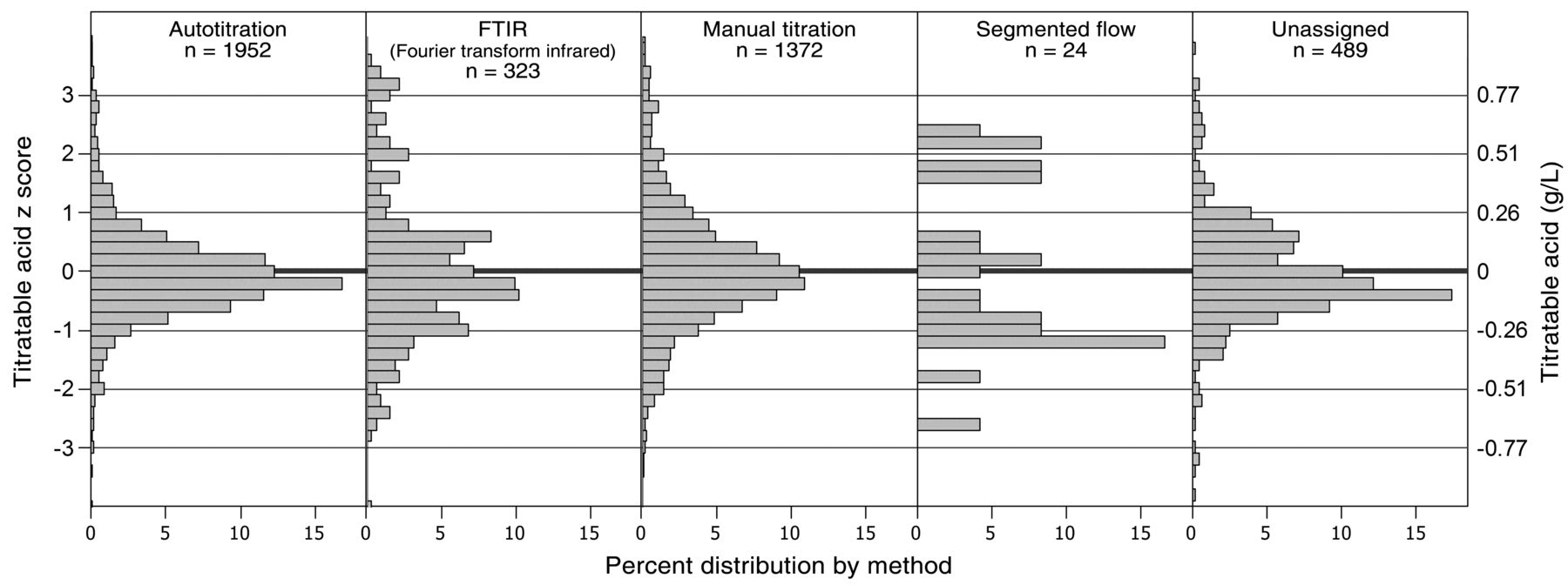
Z-score distributions of titratable acid concentrations by self-reported titratable acid analysis method. The left axis is the z-score and the right axis is equivalent units. Scores were normalized by individual wine results to allow analytical comparison across all wines.

Z-score distributions of malic acid concentrations by self-reported malic acid analysis method. The left axis is the z-score and the right axis is equivalent units. Scores were normalized by individual wine results to allow analytical comparison across all wines.

Z-score distributions of specific gravities by self-reported specific gravity analysis method. The left axis is the z-score and the right axis is equivalent units. Scores were normalized by individual wine results to allow analytical comparison across all wines.
The distillation/density method for measuring alcohol yielded values of −0.63 z (about 0.10% v/v lower than the average). Potentially, this is due to incomplete recovery of ethanol during distillation, errors in the attempering of samples, or issues with determination of mass (AOAC 2012); alternatively, these values could be the true results and the other methods are delivering high values, as explained earlier. Ebulliometry had a higher SD than other methods and its mean z-value was +0.40 (~0.06%) higher than the overall z-value. Descriptive statistics of the data show a second mode at +1.87 z (+0.3% v/v). The difference of this mode from the mean is approximately +0.24% alcohol, and may be explained by the effect of sugar on ebulliometer boiling points (Figure 6). One recommended sugar correction is to subtract 0.05 times the percent reducing sugar in the wine from the apparent ebulliometer alcohol concentration, and that this correction factor is only relevant to perform when reducing sugars are >20 g/L (Zoecklein et al. 1994). Of the wines tested, 23 had sugar concentrations >20 g/L, with an average value of 36 g/L. Using the formula above, an uncorrected ebulliometer alcohol measurement for these higher-sugar wines would be 0.2% v/v high, which accounts for most of the difference seen between the second mode and the mean. Finally, a regression of the residual sugar mean values (using residual sugar prior to cycle 19, and glucose plus fructose afterwards) against the alcohol across-lab z-scores from the self-reported ebulliometry methods gave a good fit: across-laboratory z score = 0.039 × g/L sugar − 0.0092 (n = 78, p < .01, R2 = 0.64). Since the intercept is negligible, this expression converts to: % v/v alcohol error = 0.06% RS, very close to the Zoecklein recommendation and seeming to indicate that users of the ebulliometer method are unaware of the need to correct for sugar (Figure 6 and Table 9).
Total SO2 method comparisons show that the Ripper test is biased higher with respect to aeration oxidation (2.3 mg/L) and flow injection (0.3 mg/L). However, other methods with fewer data points such as enzymatic methods (biased 8.8 mg/L higher than the average) and FTIR (biased 7 mg/L lower than the average) indicate a large variation in methods. The skew and multimodal distributions of the segmented flow and flow injected methods make direct comparisons even more challenging (Figure 7 and Table 9). Total SO2 results are expected to be independent of the method used and aside from the challenges of oxidative stability, manufacturing a certified reference material in a wine matrix may be appropriate to evaluate method accuracy. In any total SO2 method, there is a balance between maximizing the dissociation of carbonyl-bisulfite adducts (usually done at a high pH or with heat) and minimizing the oxidation of sulfites, which occurs more readily at high pH, during the analysis (Joslyn 1955). In addition to these considerations, method-specific issues, such as the ability of iodine to react with non-sulfite reducing agents, can affect accuracy (Joslyn 1955). The AOAC reference method for total SO2 in wine is the Monier-Williams method, which is similar to the more commonly used aeration oxidation method, with the primary differences in the glassware design, sample volume, gas flow rate, and selection of acidifying agent (AOAC 2012, Williams et al. 1992). Finally, the aeration oxidation method for total SO2 must be optimized to dissociate the bound adducts with acid and heat while minimizing the potential carryover of volatile acid, and also balance the gas flow rate to allow complete carryover of gaseous SO2 while allowing adequate time for reaction with H2O2 in the receiver flask (Rankine and Pocock 1970). These competing reactions and method limitations may explain why some authors have reported lower values for total SO2 with iodometric titration than with aeration oxidation (Buechsenstein and Ough 1978). While this type of bias may occur in some wine analytical labs, our large data set of 78 wines reveals that across-laboratory reproducibility for individual methods is much poorer than within-laboratory repeatability, and that singular within-laboratory comparisons are not necessarily appropriate for broader statements about methodological bias.
Among the methods to determine free SO2, the aeration oxidation, flow injection, and segmented flow methods have similar distributions and means. As with total SO2, the Ripper method (iodometric titration) results in significantly higher values than flow injection (3.7 mg/L less) and aeration oxidation (2.7 mg/L less), although all three methods are within one SD (Figure 8 and Table 9). Some of the distributions are skewed, which indicates additional bias. The higher value by Ripper than by aeration oxidation is comparable to the bias observed previously in an intra-laboratory comparison (Buechsenstein and Ough 1978), potentially due to titration of other reducing species. Although many of the challenges in free SO2 analyses are similar to those encountered with total SO2, free SO2 analyses must also minimize bisulfite adduct dissolution. In most cases, this is considered impossible; early researchers specifically warned against using aeration oxidation to determine free SO2 in red wines (Rankine and Pocock 1970) and carefully noted the correct conditions for using the Ripper for free SO2 on wines with carbonyl-bisulfite adducts (Joslyn 1955). In addition, temperature can affect the analysis, as increased temperature increases the dissociation rate of bisulfite adducts and also impacts the equilibrium of the sulfurous acid species (Rankine and Pocock 1970, Usseglio-Tomasset and Bosia 1984). It is inappropriate to discuss which method for free SO2 analysis yields the most accurate results, since it is not clear that any of the widely used methods accounts for these factors.
There is great overlap in the results from volatile acid methods, but the Cash still is biased significantly higher than most other methods (capillary electrophoresis, enzymatic, FTIR, GC, HPLC, or segmented flow) by 0.04 to 0.05 g/L as acetic. One potential issue is the target analytes of these methods: volatile acidity encompasses all volatile short chain fatty acids (formic, acetic, propionic, etc.; Amerine and Ough 1980), but capillary electrophoreses, enzymatic, GC, and HPLC methods specifically target acetic acid. When the volatile acidity distillation is properly controlled, the correlation with acetic acid in wines is 1:1, which should indicate a false dichotomy between these two terms (Dubernet and Peraldi 2006). In practice, the Cash still method can suffer from several interferences, which include sorbate, sulfur dioxide, lactic acid, and carbon dioxide (Cottrell et al. 1985, Dubernet and Peraldi 2006, Gowans 1964, Pilone 1967), which could account for the higher observed values if not well controlled. It is interesting to note the discrepancy between the Cash still method and the segmented flow method, as one of the segmented flow instruments contains a miniaturized distillation apparatus, making that method more comparable with the Cash still method. This may indicate differences in method application between the two devices. The FTIR distribution is multimodal, with a second mode at −0.06 g/L from the mean (Figure 9 and Table 9). Part of this disparity may be due the interchangeable use of the term “volatile acid” with the primary target analyte, acetic acid, especially with the FTIR, as the calibrations may be based on different primary methods. Secondarily, IR methods are not suitable for analyte concentrations less than 0.2 g/L; because of their similarity in chemical structure, acids and sugars can mutually interfere with quantification, especially if approaching the detection limit (Bauer et al. 2008). This indicates that wines with high sugar and acids could affect the precision of IR results, especially at lower volatile acidities.
The FTIR multimodality appears again with titratable acid values, with three modes at −0.03, −0.01, and +0.13 g/L from the mean (Figure 10 and Table 9). Unlike volatile acidity (which is theoretically different from acetic acid), the choice of primary method used for calibration seems less likely as an explanation. Some other factor, such as interferences or improper calibrations, might explain the distribution; again, because of the similar chemical structure of acids and sugars, IR methods are sensitive to interferences from these analytes. Manual titration shows a skew and a significantly higher result (+0.02 g/L higher than average) than autotitration, which is significantly lower (0.01 g/L lower than the average) (Figure 10 and Table 9). Common interferences in titratable acid include endpoint definition and interference of carbon dioxide, which may affect the manual results (Guymon 1963).
Malic acid data show significantly higher values by FTIR (+0.14 g/L as compared to overall mean), capillary electrophoresis (+0.12 g/L), and HPLC (+0.08 g/L) as compared to enzymatic methods (−0.1 g/L) (Figure 11 and Table 9). While it is not possible to determine which approaches are more accurate, most enzymatic methods have a large number of potential systemic errors in both instrumental variability (calibration of spectrophotometers and pipets) and methodological variability (reaction times, enzyme activity) (Henniger and Mascaro 1985), which could contribute to a consistently low result. As mentioned earlier, the reproducibility of the malic acid enzymatic methods is most compromised at low concentrations, <0.5 g/L. Also, IR methods for malic acid, especially at lower concentrations approaching the limits of the methods (0.2 g/L) are particularly sensitive to interference from other acids and from sugars (Bauer et al. 2008).
Finally, the distributions for the different specific gravity methods show bimodality in all methods except those with small samples sizes (Figure 12 and Table 9). This bimodality was first evident in the histograms of all the data combined, as discussed earlier. The average difference between the two major modes in every method was between 0.0012 and 0.0024 specific gravity units. Since the specific gravity of water at 20°C is 1.000 and the density of water at 20°C is 0.9982 (a difference of 0.0018), the most likely explanation is human operator error in confusing density and specific gravity. The mass/known volume method is biased significantly lower than the other methods (−0.0032 specific gravity units, Table 9). This method requires proper calibration of balances, thermometers, and volumetric containers, along with proper determination of the density of water, and it is not surprising that a systematic error might be involved (AOAC 2012).
From discussions with CTS, the ASEV lab proficiency ad hoc committee, and unnamed participants, it seems that many laboratories are using the collaborative service without having their own in-house quality control program in place. Such a quality control program would include method validation, use of certified reference standards, ongoing analyst training, and control samples (ISO 2005). The lack of such quality control programs industry-wide undoubtedly has a negative effect on the overall results of the CTS results, most likely inflating the values for repeatability and reproducibility, but having an unknown impact on overall performance.
Conclusion
Thirteen years of collaborative testing data indicate that many wine industry performance issues could likely be addressed by simple laboratory quality control systems, including in-house method validation, calibration schedules, and ongoing checks of volumetric and quantitative equipment such as spectrophotometers, balances, and pH meters. Such a program involves increased training and education of laboratory technicians and managers. Other data, specifically for alcohol analysis, indicate that improvements in technology can have an impact regardless of other quality considerations. Ironically, introduction of similarly new technologies for measurement of titratable acidity, pH, and malic acid has coincided with worsening precision for these analyses. Across-laboratory industry precision (reproducibility) is currently 3.6 to 57 times worse than within-laboratory precision (repeatability), depending on the analyte. Some regulated analytes, such as SO2 and volatile acidity, could benefit from an improvement in methods. Specific gravity imprecision would undoubtedly be reduced by ensuring that specific gravity rather than density is reported. Application of the Horwitz ratio (HorRat) to this data indicates that three analytes are within or near the upper limits of internationally acceptable precision: alcohol, titratable acidity, and total SO2, while other analytes have not met this precision standard: free SO2, malic acid, volatile acid, and any of the sugar measurements. Of these, analytical performance for malic acid at low concentrations (<0.5 g/L) is particularly problematic, as measurements at such concentrations are critical to evaluating completion of malolactic fermentation. Finally, although some methods yield significantly different mean values and precisions for the same analyte, it is still challenging to evaluate the relative accuracy of methods. Certified reference materials containing these analytes, as have been developed for other foodstuffs, will be necessary to determine the accuracy of individual methods or laboratories. Further analysis of this data, particularly looking at the correlation between individual method precision and wine matrix, may contribute to a more thorough understanding of the strengths or limitations of specific methods.
Acknowledgments
Special thanks to Steven Tallman, Tom Collins, and the other members of the ASEV ad-hoc Laboratory Proficiency Committee, and to Scott Granish of Collaborative Testing Service for their assistance and input. The authors are also grateful to the industry donors of wines to the CTS program for analysis.
Footnotes
Supplemental data is freely available with the online version of this article at www.ajevonline.org.
- Received October 2014.
- Revision received February 2015.
- Accepted February 2015.
- Published online December 1969
- ©2015 by the American Society for Enology and Viticulture
Literature Cited
Vol 66 Issue 3








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Chemical analysis in the wine industry
- Analytical performance terminology
- Collaborative laboratory data in the wine industry
- The Horwitz ratio (HorRat) for evaluating analytical performance
- History and description of the Collaborative Testing Services (CTS) program
- Materials and Methods
- Results and Discussion
- Conclusion
- Acknowledgments
- Footnotes
- Literature Cited
- Figures & Data
- Supplemental
- Info & Metrics
Related Articles
Cited By...
More from this TOC section
Similar Articles
