


Abstract
Conventional methods such as Ripper titration and aeration-oxidation (A-O) are widely used for the analysis of sulfur dioxide (SO2) in wine. However, the free SO2 reported by these procedures is overestimated due to dissociation of weakly bound SO2 forms during the analysis, particularly from anthocyanin-bisulfite complexes. “Truly” free SO2 in wine can be determined from the headspace SO2 concentration of an equilibrated wine sample. A headspace SO2 method based on gas detection tubes (HS-GDT) was recently described but is not readily automated. While solid phase microextraction (SPME) yielded poor precision in our experiments, our new method, based on static headspace gas chromatography and sulfur chemiluminescence detection (HS-GC-SCD), is readily automated and achieves high precision (<5%) and low limits of detection (0.033 mg/L molecular SO2, or ~1 mg/L free SO2 in wine at pH 3.5). A-O, Ripper, HS-GC-SCD, and HS-GDT methods were compared on a diverse set of wine samples. Results from HS-GC were correlated with those from the HS-GDT method (r2 = 0.92) and achieved higher precision (relative standard deviation = 3.7%). HS-GC was highly correlated with A-O in white wines (r2 = 0.85, slope = 0.90) but had weaker correlation for red wines (r2 = 0.71, slope = 0.44). The flexibility of GC for other procedures as well as its stability and low operating costs per sample make it an attractive option, and headspace methods have been shown to be better for predicting microbial stability in red wines.
Sulfur dioxide (SO2) is the oldest and arguably one of the most important additives used in winemaking. When present in sufficient concentration, SO2 has five major effects in wine/musts: (1) SO2 is a strong antimicrobial agent and provides a protection against a wide array of detrimental microorganisms; (2) it is an effective antioxidant that consumes oxidants such as hydrogen peroxide or quinones formed during the course of wine/must oxidation; (3) it can inhibit polyphenol oxidase enzymes present in grapes; (4) it reversibly binds and bleaches wine pigments, particularly monomeric anthocyanins; and (5) it reversibly binds aldehydes and ketones produced by oxidation or during fermentation, rendering them non-odorous (Waterhouse et al. 2016).
SO2 gives a weak, diprotic acid in aqueous solution (pKa1 = 1.81, pKa2 = 7.20 in H2O at 20°C) and can exist in molecular (SO2), bisulfite (HSO3−), or sulfite (SO32−) forms. In the typical pH range of wine (3.0 to 4.0), the dominant species is the bisulfite anion, which acts as an antioxidant and participates in various binding/complexing reactions. Molecular SO2, the main antimicrobial form of SO2, is present at only a small fraction (<5%) of the HSO3− concentration at wine pH. SO32− is present at even a smaller fraction of the HSO3− concentration (<0.1%) at wine pH; thus, its influence on wine stability is likely negligible. SO2 in wine is further divided into two classes: free and bound. Free SO2 is defined as the sum of molecular and bisulfite forms and is the class with antimicrobial, antioxidant, and enzyme-inhibiting properties. Bound SO2 comprises the bisulfites that react (both weakly and strongly) with other molecules within the wine matrix and do not exhibit those protective properties, with some exceptions (Wells and Osborne 2011). The sum of the free and bound sulfites defines the “total” sulfite concentration (Buechsenstein and Ough 1978).
To obtain enologically useful information, analytical methods for SO2 must distinguish between the free form with its protective properties and the bound forms, which do not have these properties. Common analytical methods for free SO2 in wineries include iodometric titration (Ripper method) and aeration-oxidation (A-O) method (Iland et al. 1993, Urbano-Cuadrado et al. 2004). These standard methods utilize an initial acidification step to avoid interferences from phenolics (Ripper) or to favor the molecular SO2 species prior to a separation step (A-O and related flow injection or segmented flow analysis methods). This acidification step, coupled with consumption of free SO2 during the course of analysis, can result in release and subsequent measurement of weakly bound SO2, particularly from anthocyanin-bisulfite complexes. As a result, standard measurement approaches will overestimate free SO2, particularly in red wines (Coelho et al. 2015).
This artefactual overestimation of free SO2 can be avoided by measuring the headspace SO2 concentration of an equilibrated wine sample. This concentration of headspace SO2 can be related to the aqueous molecular SO2 concentration by its Henry’s Law coefficient (H), which can then be related to the concentration of “truly” free SO2 by pKa1 and the Henderson-Hasselbalch equation. To calculate free SO2, the ethanol concentration, pH, and temperature of the wine sample must be accurately known to establish the correct values of pKa1 and H. A recently described approach used a syringe to create an equilibrated enclosed headspace above a wine sample and then expel the headspace through a commercial gas detection tube (GDT). The GDTs contain a colorimetric SO2-selective reagent such that length of discoloration on the GDT is proportional to the analyte concentration. The HS-GDT technique does not involve pH shifts, sample dilution, or temperature changes, and thus avoids disturbances in SO2 equilibria in wine or contributions from weakly bound SO2 (Coelho et al. 2015). The authors observed that A-O resulted in an approximately three-fold overestimation of free SO2 in red wines compared to HS-GDT. The HS-GDT approach was later shown to yield more accurate predictions of yeast survivability and viability during challenge tests, suggesting that “truly” free SO2 measurements may be of greater relevance for prediction of antimicrobial activity (Howe et al. 2018).
Although easy to implement, one drawback of the HS-GDT approach is that it is not readily automated. Other potentially more automatable approaches for indirect and direct measurement of “truly” free SO2 have been described, including capillary electrophoresis (CE) and headspace gas chromatography (HS-GC) coupled to an electrolytic conductivity detector (ECD). These studies have come to similar conclusions that free SO2 may be overestimated by up to an order of magnitude in red wines, although none of the methods appear to be widely used (Davis et al. 1983, Boulton et al. 1996, Collins and Boulton 1996).
The coupling of HS-GC with a sulfur chemiluminescence detector (SCD) for analysis of SO2 and other volatile sulfur compounds in unadjusted wine samples was recently described (Ontanon et al. 2019). HS-GC-SCD is readily automated and has excellent selectivity for sulfur compounds. Because samples were not adjusted or heated prior to or during analysis, the SO2 measured by HS-GC-SCD should be proportional to the “truly” free SO2. However, this earlier report did not compare results for wines analyzed by HS-GC-SCD to those analyzed by other analytical approaches for measuring SO2. In this work, we reported development of an HS-GC-SCD method for “truly” free SO2, and compared it to other methods for measuring SO2 (HS-GDT, A-O, Ripper). We also compared the differences between headspace methods and conventional methods (A-O, Ripper) for measurement of substances that might form metastable bound SO2 that could be released during conventional analyses to evaluate a basis for the discrepancy between the headspace and conventional methods.
Materials and Methods
Chemicals
Potassium metabisulfite (97%); acetaldehyde (99%); 2-ketoglutaric acid (99%); pyruvic acid (99%); 2,4-di-nitrophenylhydrazine (DNPH); ammonium dihydrogen phosphate (≥95%); formic acid (≥95%); methanol (≥99.9%); and acetonitrile (≥99.9%) were obtained from Sigma-Aldrich. L-Tartaric acid (99%) was obtained from Fisher Scientific. Ethanol (anhydrous, ≥99.5%) was obtained from Decon Laboratories. Hydrogen peroxide (30% w/v), sodium hydroxide (10%, 0.1N, and 0.01N), o-phosphoric acid (85%), sulfuric acid (25%), starch (1%), and iodine (0.02N) were obtained from Enartis Vinquiry. Ethyl methyl sulfide (1000 μg/mL) was obtained from SPEX CertiPrep.
SO2 working standards
SO2 stock solutions at nominal concentrations of 6000 mg/L as SO2 were prepared weekly by dissolving potassium metabisulfite in a 10% (v/v in water) solution of methanol to avoid SO2 autooxidation. Working standards were then prepared as needed by adding an appropriate volume of a stock SO2 solution to model wine. Model wine solution was prepared in ultrapure water containing 4 g/L of tartaric acid and 10% ethanol and adjusted with NaOH solution to a pH of 3.50. The ethanol concentration was verified using an Alcolyzer Wine M. The true pKa (pKM) for SO2 in each batch of model wine was determined using the following calculations, and the concentration of each of the calibration standards was calculated using the Henderson-Hasselbalch equation.
Estimation of pKa1 (pKM) of SO2 and calculation of free SO2 from molecular SO2
The following equations were built from a multiple linear regression model using XLSTAT (Add-insoft) to predict the pKa values contained in published tables (Usseglio-Tomasset and Bosia 1984).
To estimate the value of the thermodynamic constant pKT for various alcohol concentrations (Alc., %v/v) and temperatures (T, °C), the following equation was used (Equation 1).
Estimation of pKT.
 Eq. 1
Eq. 1
To estimate the value of the coefficients A and B for various alcohol concentrations and temperatures, the following two equations were used (Equations 2 and 3).
Estimation of A constant.
 Eq. 2
Eq. 2
Estimation of B constant.
 Eq. 3
Eq. 3
Finally, the value of the mixed dissociation constant, pKM, as a function of pKT, the coefficients A and B, and the ionic strength (I) was determined by Equation 4 below.
Estimation of pKM.
 Eq. 4
Eq. 4
Because the measurement of ionic strength (I) is complex and labor intensive, a typical ionic strength of 0.056 M can be assumed (the typical range for ionic strength in wine is 0.016 M to 0.100 M) and was used in the calculation of pKM (Berg and Keefer 1958, 1959, Ough et al. 1982, Abgueguen and Boulton 1993) without resulting in significant error in estimation of free SO2 (Coelho et al. 2015).
The value of pKM can then be used in the Henderson-Hasselbalch equation (Equation 5) to determine the molecular and free species of SO2 as a function of pH.
Modified Henderson-Hasselbalch equation.
 Eq. 5
Eq. 5
SO2 measurements using previously described approaches: A-O, Ripper, and HS-GDT
SO2 analysis by A-O (Iland et al. 1993), Ripper (Vahl and Converse 1980), and HS-GDT (Coelho et al. 2015) were all performed in triplicate for each wine. The Ripper method was also used to measure total SO2.
SO2 measurement by HS-GC-SCD
Analysis of molecular and free SO2 were performed with an Agilent 7890B gas chromatograph coupled with an Agilent 8355 SCD (Agilent Technologies). The capillary column used was an Agilent DB-WAX-UI (30 m × 0.25 mm i.d. × 0.25 μm film thickness). The autosampler was a PAL3 RSI from CTC analytics operated in static headspace mode. The 2.5-mL gas-tight syringe was heated to 40°C to prevent condensation of the headspace sample in the syringe. Injections were split (4:1 ratio) at an injector temperature of 200°C. Before and after injection, the syringe was purged with pure He for 90 sec. The temperature program for the final method started at 50°C, which was maintained for 2.5 min, increased at a rate of 50°C/min to 220°C, and held at this temperature for 2 min. The complete chromatogram took 7.9 min, with a total GC cycle time of 10.5 min between injections. The carrier gas was He (44.2 cm/sec) in constant flow mode. The SCD burner temperature was 800°C with a hydrogen flow rate of 100 mL/min and an air flow rate of 40 mL/min. The SCD pressure was 6 Torr with the controller at 200 Torr.
Immediately after opening a bottle, 15 mL of room temperature wine (23°C) was transferred into a 20-mL amber crimp top headspace vial and spiked with 50 μL of internal standard (30 μg/mL ethyl methyl sulfide in methanol) for each analysis. The vials were then capped with magnetic crimp seals with PTFE/silicone septa. If not already equilibrated to room temperature, the samples were equilibrated for 1 hr before running the procedure. HS-GC-SCD analyses were then performed as described above. Because a headspace volume of 50 mL at room temperature has already been shown to take a minimum of 5 min to fully equilibrate, the equilibration time for the GC vials with 5 mL of headspace volume was assumed to be equivalent or less (Coelho et al. 2015). The analytical characteristics of the method are summarized in Table 1.
Method figures of merit.
Monomeric anthocyanins by high performance liquid chromatography (HPLC)
The separation of the monomeric anthocyanins was conducted with reverse-phase HPLC using an Agilent 1100 series (Agilent Technologies) modular HPLC system based on the method described elsewhere (Ritchey and Waterhouse 1999). The HPLC system included a system controller, G1379A degasser, G1311A quaternary pump, G1313A autosampler, G1316A column compartment, and a G1315A DAD/UV-vis detector. Data was processed using ChemStation version B.04. Separation of anthocyanins was performed with a LiChrospher 100 RP-18 column (4 × 250 mm, 5 μm particle size; Agilent Technologies). A guard column of the same material was also installed, and column temperature was maintained at 40°C.
Briefly, the procedure used two mobile phase solutions for analysis. The solvents were (A) 50 mM ammonium dihydrogen phosphate (Sigma-Aldrich ≥95%) adjusted to pH 2.6, and (B) 20% Mobile A + 80% acetonitrile (v/v) (Sigma-Aldrich ≥99.9%). The gradient used was: zero-time conditions were 94% A and 6% B; the pumps were adjusted to 70% A and 30% B at 15 min; to 50% A and 50% B at 30 min; to 40% A and 60% B at 35 min; and to 94% A and 6% B at 41 min (end of analysis). After a 10-min equilibrium period, the next sample was injected.
The concentration of total monomeric anthocyanins was determined by the summation of the peak areas measured at 520 nm for delphinidin 3-glucoside, pelargonidin, cyanidin 3-glucoside, pelargonidin 3-glucoside, delphinidin, malvidin 3-glucoside, and malvidin. The concentration was expressed as mg/L of malvidin-3-glucoside equivalents.
Free and SO2-bound wine carbonyls by HPLC
Acetaldehyde, 2-ketoglutarate, and pyruvate were determined by HPLC after derivatization reaction with DNPH reagent (Sigma-Aldrich) as reported (Han et al. 2015). Briefly, aliquots of wine samples (100 μL) were dispensed into a vial, followed by the addition of 20 μL of freshly prepared 1120 mg/L SO2 solution, 20 μL of 25% sulfuric acid, and 140 μL of 2 g/L DNPH reagent. After mixing, the solution was allowed to react for 15 min at 65°C and then was promptly cooled to room temperature in a water bath. Carbonyl hydrazones were analyzed by HPLC using the system described above. In the chromatographic system, a ZORBAX Rapid Resolution HT, SB-C18 column (1.8 μm, 4.6 × 100mm2; Agilent Technologies) was used for separation. Separation was obtained using a flow rate of 0.75 mL/min and column temperature of 35°C, and the mobile phase solvents were (A) 0.5% formic acid (Sigma Aldrich ≥95%) in water milli-Q and (B) acetonitrile (Sigma Aldrich ≥99.9%). The gradient elution protocol was as follows: 35% B to 60% B (0 to 8 min); 60% B to 90% B (9 to 13 min); 90% B to 95% B (14 to 15 min, 2-min hold); and 95% B to 35% B (16 to 20 min, 4-min hold), with a total run time of 20 min. Eluted peaks were measured at 365 nm and were compared with derivatized acetaldehyde, 2-ketoglutarate, and pyruvate standards (Sigma-Aldrich).
Analysis of alcohol, pH, and temperature
Alcohol
The ethanol content of all wine samples and model wines was determined using an Alcolyzer Wine M (Anton-Paar).
pH
The pH of all wine samples and model wines was measured using an Orion 5 Star (Thermo Scientific). The pH probe was calibrated daily using buffers of 2.00, 4.01, and 7.00 pH standards. Slopes of each calibration ranged from 96 to 100%.
Temperature
Sample temperature was measured using VWR Traceable Lollipop Water-Resistant Thermometers.
Wine samples
Table 2 shows the identity of the wines used to compare the four methods. Various wines (n = 27) covering a range of varieties, vintages, and appellations were donated from Constellation Brands.
Wines used for the comparison of methods and respective sample codes.
Results and Discussion
In our initial work, we evaluated the use of solid-phase microextraction (SPME) followed by separation on a porous layer open tubular (PLOT) GC column, since similar methods have been used for analysis of other volatile sulfur compounds in wine. This initial approach used short SPME exposure times to avoid perturbation of equilibria but was determined to be unacceptable due to poor precision and excessive peak broadening on the PLOT column that was difficult to analyze (data not shown). We then evaluated static headspace injection with different columns (DB-Sulfur, DB-WAX-ETR, and DB-WAX-UI). We selected the DB-WAX-UI column because it could achieve rapid separation of SO2 with gaussian peak shape, excellent peak precision (2.8% relative standard deviation [RSD]), and a low limit of detection. The final GC parameters used were similar to a reported method, with the exceptions of eliminating the SO2 preconcentration step in favor of drawing a 0.500-mL sample directly from the headspace vial with the autosampler and using ethyl methyl sulfide (EMS) as the internal standard (Carrascon et al. 2017). By design, the use of an SCD as opposed to a mass spectrometer detector was intended to improve sensitivity and selectivity to SO2. The selected column does degrade with time from the SO2 exposure and should be replaced after ~200 injections.
With the DB-WAX-UI column and corresponding GC parameters for this method, the elution time for the SO2 peak and EMS internal standard was ~3.2 and 1.8 min, respectively, and neither of these co-eluted with any other potentially interfering compounds typically found in wine. A representative chromatogram of a 2014 Central Coast Viognier is shown in Figure 1.

Chromatogram of a 2014 Central Coast Viognier. Column: DB-WAX-UI. Sampling method: Static headspace. DMS, dimethyl sulfide; EMS, ethyl methyl sulfide; ISTD, internal standard (EMS).
Table 3 presents free SO2 (mg/L) on a set of California wines as measured by the A-O, Ripper, HS-GDT, and HS-GC methods. The results of the A-O and Ripper methods will be referred to as “apparent” free SO2, and the free SO2 measured using the HS-GDT and HS-GC techniques will be referred to as “truly” free SO2. All analyses on each wine using each method were performed in triplicate to assess and compare the precision of the methods.
Results of free SO2 in the test wines using aeration-oxidation (A-O), Ripper, headspace gas chromatography (HS-GC), and headspace gas detection tube (HS-GDT) methods.a
Table 4 tabulates other basic chemistry parameters of the wine samples analyzed in this set. The estimates of true pKa based on alcoholic strength, temperature, and ionic strength are also shown. For SO2 measured by HS-GC, the formula for estimating the truly free SO2 is based on the Usseglio-Tommaset calculations (Usseglio-Tomasset and Bosia 1984). For SO2 measured by HS-GDT, a related approach was used in Coelho et al. (2015) for estimating the true pKa.
Standard enological data and calculated pKa values on the tested wines.
Because the temperatures of the HS-GC and HS-GDT analyses were not controlled beyond the prevailing ambient room temperature (18 to 21°C), a few comparative analyses were conducted at non-equivalent temperatures. Specifically, analysis of the 2015 Paso Robles Blaufränkisch (BLAU), 2015 California Cabernet Sauvignon (CAB), and 2014 Napa Valley Chardonnay (CHA 1) by HS-GC and HS-GDT was at a 2°C differential, with the HS-GC analysis performed at 20°C and the HS-GDT analysis performed at 18°C. Analysis of the 2014 Central Coast White Blend (WHITE), 2015 Central Coast Viognier (VIO 2), and 2014 Central Coast Chardonnay (CHA 3) samples also occurred at a 2°C differential, with the HS-GC analysis performed at 25°C and the HS-GDT analysis performed at 23°C. While the respective formulas for calculating the true pKa have built-in functions that account for the difference in temperature, it is unclear whether these are sufficient to overcome instances when analysis is done at a non-standard temperature or can account for slight variations in analysis temperature beyond ± 1°C. Given the uncertainty, these data points were excluded from the statistical analysis.
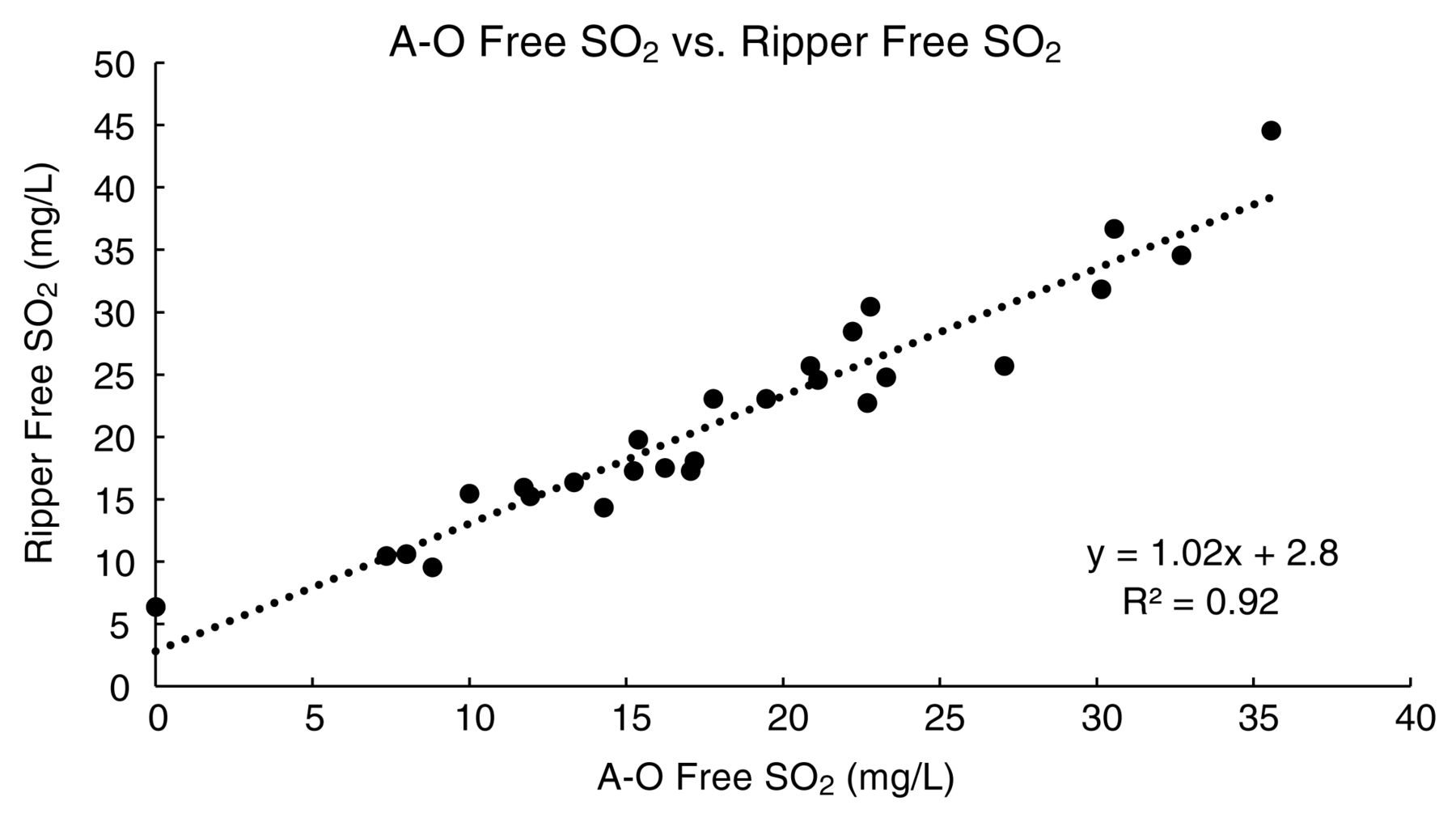
The full comparison of analytical methods indicated that analytical precision of the A-O and Ripper methods were comparable and satisfactory, as both methods had RSDs (%RSD) below 5%. Both the A-O and Ripper methods had similar average standard deviations across the 27 wines analyzed (0.8 and 0.7 mg/L free SO2, respectively). A graphical comparison of the free SO2 results of the 27 wines by A-O and Ripper analysis is shown in Figure 2. The methods showed good agreement based on a regression analysis (slope = 1.02, intercept = 2.8, r2 = 0.92, Figure 2). The highest standard deviation in free SO2 measurement by A-O was observed in the analysis of the non-vintage Brut sparkling wine, which was likely due to dissolved CO2 that carried over into the peroxide trap used in the A-O procedure and resulted in an over-titration and subsequent over-reporting of apparent free SO2. Interestingly, the overall RSD for the Ripper method (3.79%) was lower than that for the A-O method (4.60%). This finding is in contrast to findings from older studies that reported RSDs as high as 9.5 to 12% for the Ripper method (Buechsenstein and Ough 1978, Vahl and Converse 1980), but in agreement with more recent results from interlaboratory proficiency testing that observed little variation in precision between the two methods (Howe et al. 2015). The average absolute difference in free SO2 between the two methods was 3.3 mg/L, with the maximum absolute difference 9.0 mg/L. In most cases, the free SO2 results measured by Ripper were 0.7 to 5.8 mg/L higher than the free SO2 measured by A-O. This effect may be due to over-titration beyond the true end point by the operator to reach a visually detectable end point, especially in darkly pigmented samples, or due to the presence of interfering compounds such as reducing sugars or ascorbic acid (Iland et al. 1993).

Correlation of free SO2 values measured by aeration-oxidation (A-O) and Ripper methods.
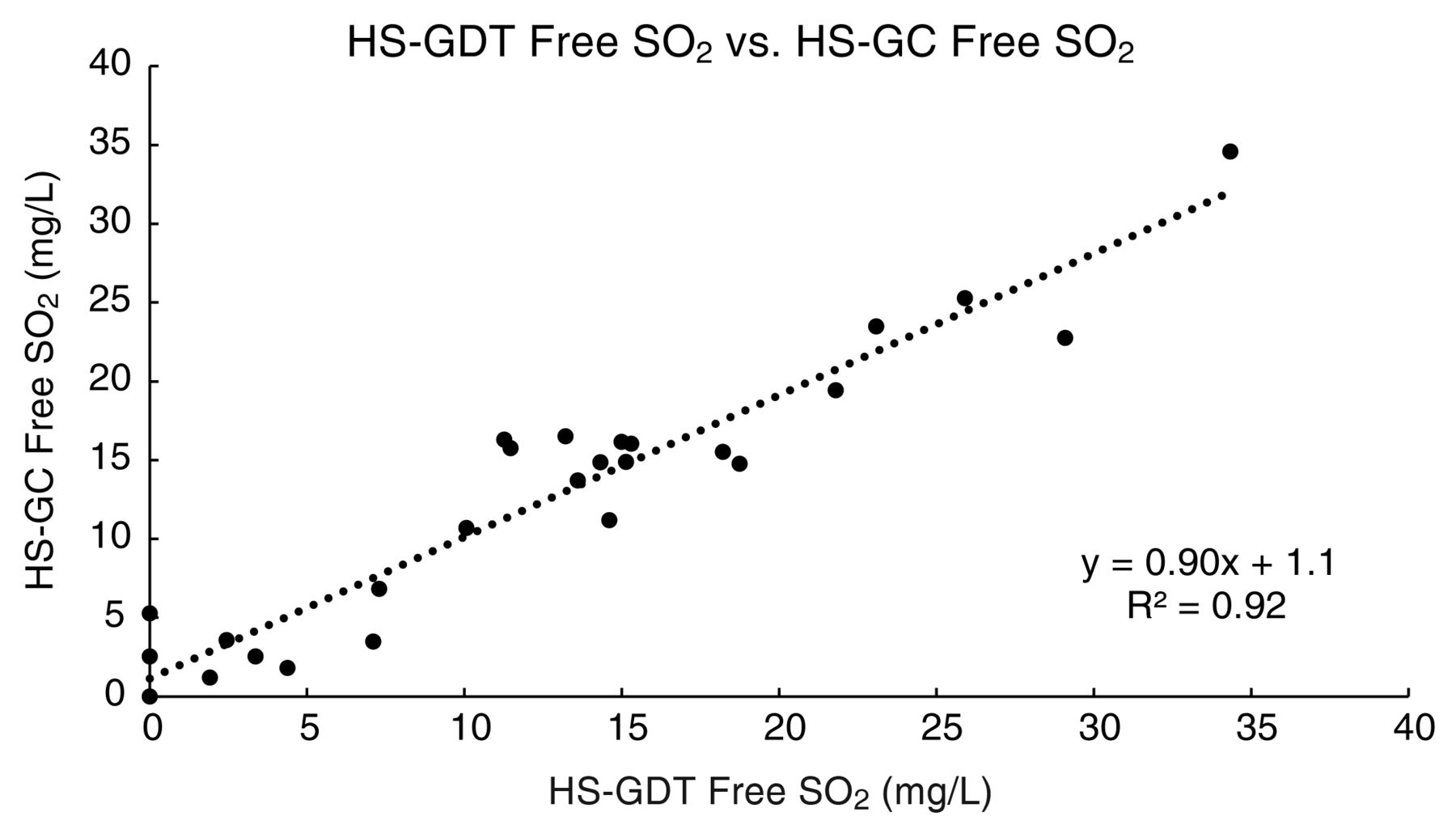
With respect to the headspace techniques for measuring free SO2 (after mathematical conversion from molecular SO2), good linear agreement was observed between the HS-GC and HS-GDT methods (slope = 0.90, intercept = 1.1, r2 = 0.92, Figure 3). The average absolute difference in free SO2 between the two methods was 2.1 mg/L of free SO2, with a maximum absolute difference of 6.3 mg/L. The HS-GC and HS-GDT methods had average standard deviations of 0.4 and 1.4 mg/L free SO2, respectively. In terms of analytical precision, the HS-GC technique had an RSD (3.72%) that was appreciably lower than the RSD for the HS-GDT method (11.83%). The lower precision of the HS-GDT method was likely due to the difficulty in reproducibly identifying the start and stop points of tube staining.
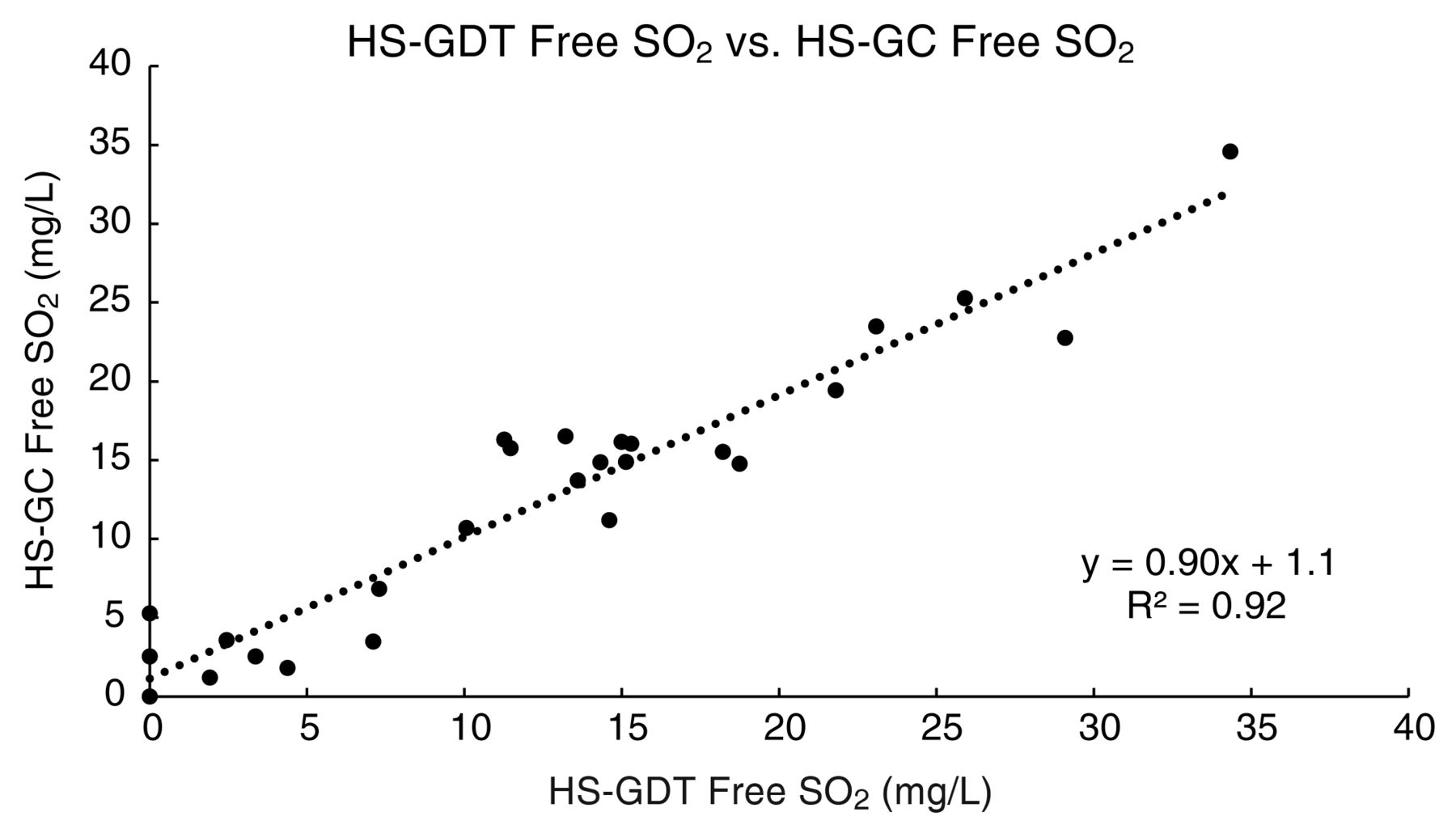
Correlation of free SO2 values measured by the headspace gas detection tube (HS-GDT) and headspace gas chromatography (HS-GC) methods.
Since partitioning of SO2 in the headspace is governed by Henry’s Law, effort was made to ensure that analysis of wines by the HS-GC and HS-GDT methods was performed at the same temperature ± 1°C. The bottled wine samples were equilibrated at room temperature (23°C) for a minimum of 24 hrs prior to analysis. Temperature of the wine samples was recorded at the time of each batch of HS-GDT analysis. For the HS-GC analysis, the heating element of the sample agitator was turned off because precise temperature control was not available under 30°C; therefore, samples in the GC vials were at the prevailing room temperature at the day and time of analysis. The laboratory is temperature controlled within 2 to 3°C for comfort but is not regulated to ± 1°C. Despite those efforts, the difference in results between the HS-GC and HS-GDT methods could be due to slight differences (>1°C) in analysis temperatures. Moreover, the imprecision of the end point determination with the GDTs may have amplified the apparent differences. Better temperature control should be possible to improve the correlation and precision. In practice, precise management of GDT temperatures would be difficult in a small-scale operation.
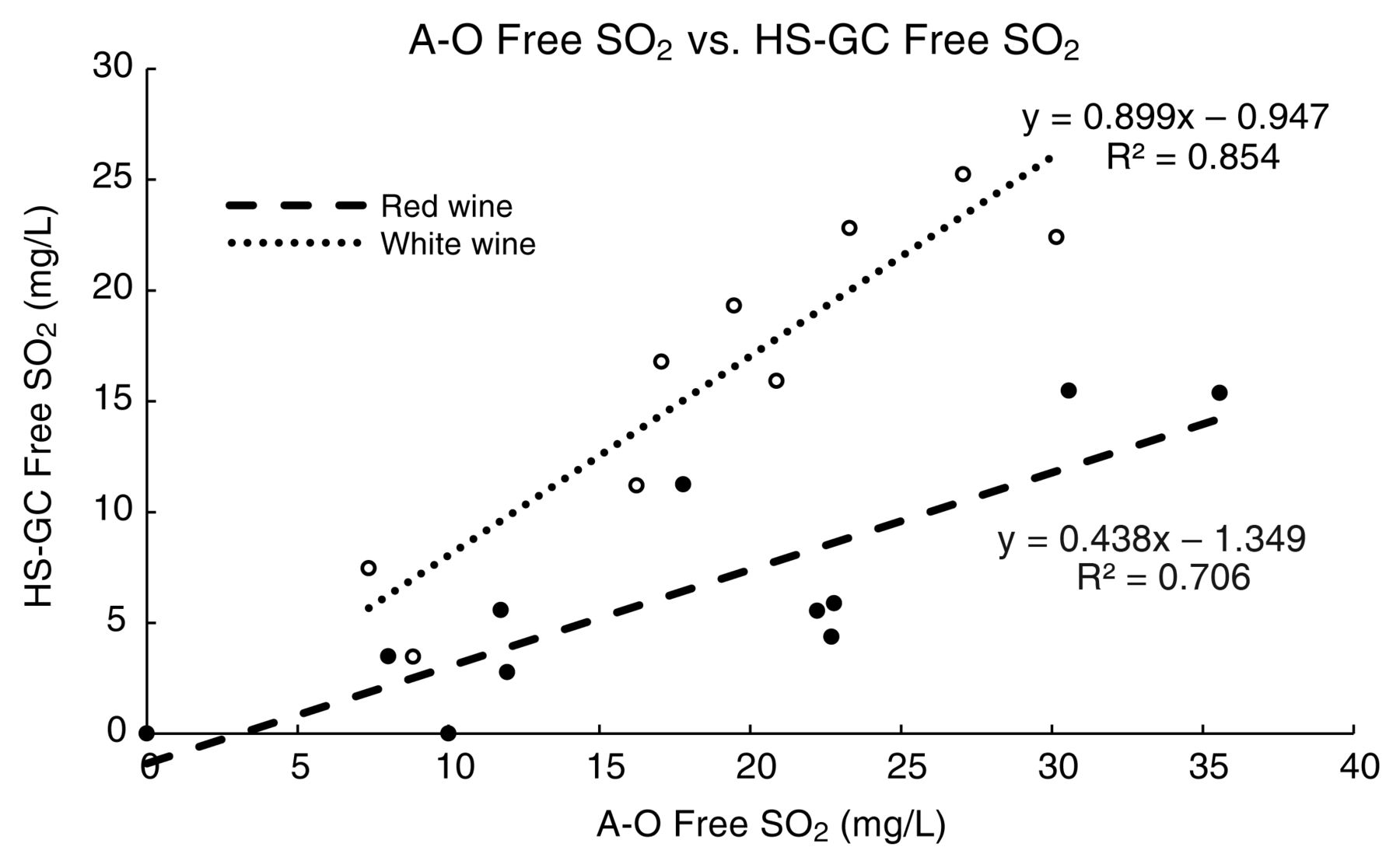
For red wines, measured free SO2 was higher with the A-O method than with the HS-GC method (range 5 to 20 mg/L, Table 3). On average, HS-GC free SO2 values for red wines were only 39% of those measured by A-O values (61% lower). For white wines, better agreement in free SO2 values was observed between the HS-GC and A-O methods. On average, the free SO2 values for white wines measured by the HS-GC method were 87% of the free SO2 values (13% lower) determined by the A-O method for the same wines. Correlation between the HS-GC and A-O methods were also better for white wines (r2 = 0.85, Figure 4) than for red wines (r2 = 0.71, Figure 4). These results are comparable to previous work comparing HS-GDT and A-O, which reported that the values with HS-GDT were 51% and 13% lower for red and white wines, respectively (Coelho et al. 2015).

Correlations between aeration-oxidation (A-O) and headspace gas chromatography (HS-GC) methods for red and white wines.
To determine the possible magnitude of the error contributed by the volatilization of SO2 into the 5 mL of headspace in the amber headspace vial, the following calculations were performed.
To estimate KH as a function of temperature (in °C), the following equation (6) was used, which is the temperature correction for Henry’s Law volatility constant KH:
 Eq. 6
Eq. 6
For example, for a liquid concentration 1.8 × 10−5 M molecular SO2 at 23°C, the KH value is 1.38 M/atm. Using Henry’s Law, the vapor pressure of SO2 above the liquid would be 1.3 × 10−5 atm. The concentration of SO2 (in g/L) in the headspace is calculated using the following equation (7) and the known vapor pressure.
Calculation of headspace SO2 concentration at equilibrium.
 Eq. 7
Eq. 7
Further calculations showed that under these conditions, ~1% of the SO2 in the sample was present in the 5 mL of headspace in the GC vial that contained 15 mL of sample, a small fraction that should not significantly disrupt the free SO2 equilibrium.
To evaluate the hypothesis that the discrepancies between the two analysis methods (HS-GC and A-O) could be explained by dissociation of metastable bisulfite complexes during analysis, the 27 wines used in the study were analyzed for the concentrations of major SO2 binders, including monomeric anthocyanins, acetaldehyde, pyruvate, and 2-ketoglutarate, which are all candidate compounds for metastable bisulfite complexes (Table 5). Monomeric anthocyanins were evaluated by HPLC and expressed as mg/L of malvidin-3-glucoside equivalents. Concentrations of acetaldehyde, pyruvate, and 2-ketoglutarate in the wine samples were determined by HPLC after derivatization reaction with DNPH reagent (Han et al. 2015). We also calculated “metastable bisulfite” as the difference between the A-O and HS-GC methods and performed linear regressions for metastable SO2 binders (monomeric anthocyanins acetaldehyde, pyruvate, and 2-ketoglutarate) against the concentration of metastable bisulfite complexes observed in each wine.
Evaluation of metastable bisulfite complexes, total monomeric anthocyanins (mg/L malvidin-3-glucoside equivalents), acetaldehyde, 2-ketoglutarate, and pyruvate on California wines (n = 27).
We observed a significant correlation between monomeric anthocyanins and metastable bisulfite (r2 = 0.42, Figure 5). No correlation was observed between metastable bisulfite complexes and acetaldehyde, alpha-ketoglutarate, or pyruvate (r2 < 0.1; plots not shown) in red or white wines, suggesting that these compounds were not related to the discrepancies between methods, similar to findings reported by others (Bisson 1999). Similar correlations were observed to explain the discrepancies in measurements using the Ripper and HS-GDT techniques (data not shown). Anthocyanin-bisulfite complexes likely contribute to A-O and Ripper measurements of free SO2 due to their rapid dissociation (first-order rate constant for the dissociation of anthocyanin-bisulfite adducts = 0.2/min) (Brouillard and Elhagechahine 1980). By comparison, the first-order rate constant glucose-bisulfite complex dissociation is 3.7 × 10-4/min (Vas 1949). Some derived anthocyanin pigments that are known to also bind SO2 or respond to pH changes (Zimman and Waterhouse 2004) are not quantified by the method presented here. As demonstrated in Coelho et al. (2015), a higher correlation would likely have been found if a different method, such as SO2 bleaching, had been used for total anthocyanin content.
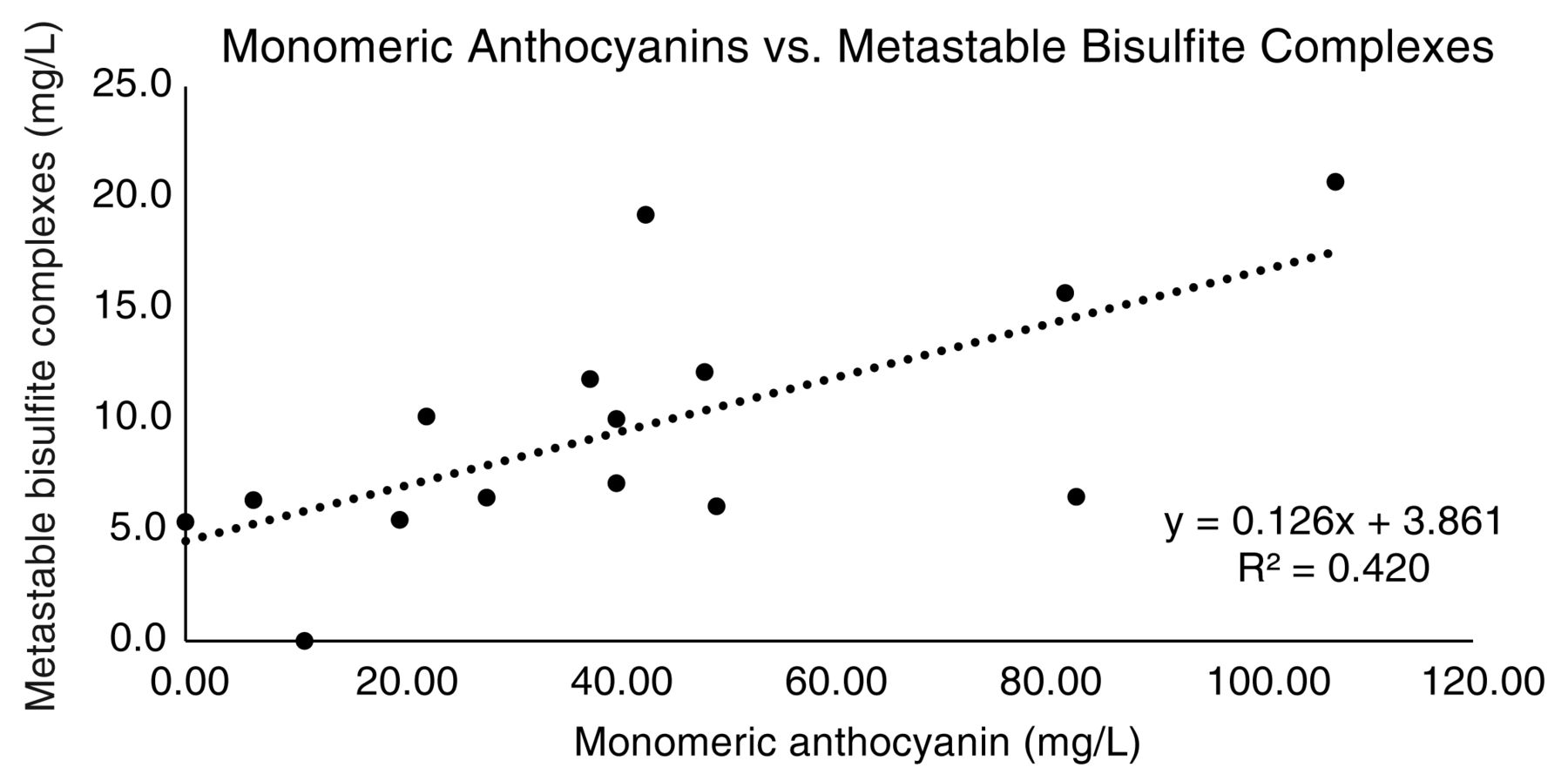
Correlation between metastable bisulfite complexes and anthocyanin concentration (red wines only). Metastable bisulfite complexes were calculated as the difference between free SO2 by aeration-oxidation (A-O) and free SO2 by headspace gas chromatography (HS-GC).
To determine the limit of detection and quantification, model wine solutions containing known trace amounts of molecular SO2 were analyzed with the described HS-GC-SCD method. The signal to noise ratios of each of the SO2 peaks were determined using the ChemStation software (version C.01.07 SR2 [255]). Limit of detection was calculated as the amount of molecular SO2 required to attain a signal to noise ratio of 3, and the limit of quantification was calculated as the amount of molecular SO2 required to attain a signal to noise ratio of 10. For the HS-GC-SCD method, the limits of detection and quantification were 0.033 mg/L and 0.067 mg/L molecular SO2, respectively. The similar HS-GC-SCD method published in Ontañón et al. (2019) reported an even lower limit of detection (0.46 μg/L molecular SO2); however, it is not clear how these values were calculated, which makes direct comparison difficult.
The increased significance of headspace versus conventional methods with regard to microbial stability in winemaking has been demonstrated (Howe et al. 2018). Conventional methods detected significant molecular SO2 that should suppress yeast, but no suppression was observed in red wine. By contrast, the headspace method properly predicted suppression at ~0.8 mg/L molecular SO2.
Conclusion
Based on a gas detection tube method, we developed an analytical procedure using HS-GC coupled with SCD that can rapidly and precisely quantify molecular and free SO2 in wine. The method requires minimal sample preparation and involves no chemical reagents (with the exception of a trace internal standard). At room temperature (23°C), the method can successfully detect levels of molecular SO2 at concentrations as low as 0.033 mg/L. The total chromatographic time for the method is eight minutes and, provided that information on the alcohol concentration and pH is readily available, the molecular and free SO2 concentrations for the sample can be rapidly calculated using simple formulae. The HS-GC method offers a high degree of precision, with a coefficient of variation of 3.72%.
In comparing SO2 analysis methods on a large set of wine samples, the HS-GC method further confirms that conventional SO2 methods systematically overestimate the molecular and free SO2 in red wines, largely due to the presence of anthocyanins. It appears that the presence of anthocyanins in wine leads to formation of metastable complexes with bisulfite that are inadvertently released during conventional analysis methods, leading to inflated and misleading results. Since headspace analysis of SO2 in wine has been shown to predict microbial stability better than conventional methods, the adoption of headspace-based methods may improve prediction of wine stability.
Acknowledgments
The authors gratefully thank Constellation Brands, St. Helena, CA, for their collaboration and generous donation of wine samples. Additional wine samples were donated from The Wine Group, Livermore, CA. Funding for this project was provided by the American Vineyard Foundation and from the Henry A. Jastro Shields Graduate Research Award. The authors declare no conflicts of interest.
Footnotes
By downloading and/or receiving this article, you agree to the Disclaimer of Warranties and Liability. The full statement of the Disclaimers is available at http://www.ajevonline.org/content/proprietary-rights-notice-ajev-online. If you do not agree to the Disclaimers, do not download and/or accept this article.
- Received July 2019.
- Revision received December 2019.
- Accepted February 2020.
- Published online July 2020
- © 2020 by the American Society for Enology and Viticulture
Literature Cited
Vol 71 Issue 3









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
More from this TOC section
Similar Articles
