
Abstract
Genome-wide association studies with SNP markers are expected to allow identification of genes that underlie complex disorders. Hundreds of thousands of SNP markers will be required for comprehensive genome-wide association studies. The development of microarray-based methods for SNP genotyping on this scale remains a demanding task, despite many recent advances in technology for the production of high-density microarrays. A key technical obstacle is the PCR amplification step, which is required to reduce the complexity of and gain sufficient sensitivity for genotyping SNPs in large, diploid genomes. The multiplexing level that can be achieved in PCR does not match that of current microarray-based methods, making PCR the limiting step in the assays. Highly multiplexed microarray systems for SNP genotyping have recently been developed by combining well-known reaction principles for DNA amplification and SNP genotyping in clever ways. These new methods offer the potential of genome-wide SNP mapping of genes involved in complex diseases in the foreseeable future, provided that issues related to selection of the optimal SNP markers, sample throughput and the cost of the assays can be addressed.
Similar content being viewed by others

Main
It has been estimated that the human genome contains more than 10 million nucleotide positions that have common (defined as >1%) variation between individuals in a population1. The prevailing interest in the large-scale genotyping of SNPs originates in the proposal that genome-wide association studies with SNP markers may enable the identification of genetic variation that predisposes to complex disorders2,3. As a result of the International Haplotype Mapping Project4 and recent data from Perlegen Sciences5, public databases currently contain data for more than 2 million SNP markers with verified allele frequencies. The aim of the International Haplotype Mapping Project is to characterize linkage disequilibrium (LD) patterns across the genome to facilitate selection of the most informative subsets of 'tagging' SNPs6 for genome-wide association studies. Studies on LD patterns suggest that genome-wide association studies will require the genotyping of several hundred thousands of SNPs in each individual7. Fine mapping of genes associated with disease in large genomic regions previously defined by linkage analysis also requires the genotyping of hundreds or thousands of SNPs. Efficient and cost-effective SNP genotyping methods will be required for routine clinical applications once disease-predisposing genes have been identified and the allelic variants that predict disease or improve diagnostics have been specified. Moreover, many of the complex diseases and traits may be caused by rare alleles, which can only be detected by resequencing complete genomic regions in multiple individuals first for identification of the variants8 and then for disease diagnostics. Thus, there are many calls for the genotyping of SNPs on a large scale.
The possibility of highly multiplexed analyses combined with the low costs per data point offered by microarray-based assays denote considerable advantages for SNP genotyping on a large scale. During the past ten years, microarray technology for the genome-wide expression analysis of tens of thousands of genes in each sample, together with computational tools for analysis of the large amount of data generated by this technology, has matured9. Analogously to expression profiling, microarray-based methods have been successfully adapted to the analysis of genomic copy-number alterations by hybridization to probes spanning large chromosomal regions using comparative genomic hybridization10. Development of robust microarray technology for genotyping at the resolution of single nucleotides on a genome-wide scale has, for reasons discussed below, proved to be a challenging task. Not until recently was there any progress in the development of microarray systems for highly multiplexed genotyping that has the potential for genome-wide SNP genotyping.
PCR strategies enabling multiplex SNP genotyping
Identification of a specific single-base change among the 3 billion bases that constitute the human genome is a challenging task, which was facilitated 20 years ago by PCR11,12. PCR offers a means of reducing the complexity of the genome and of increasing the copy number of the DNA templates to levels required for the specific and sensitive detection of single-base changes. But the design of robust PCR assays with multiplexing levels exceeding 10–20 amplicons has proved to be more difficult than initially anticipated. The reason is that, in multiplex PCR, the number of undesired interactions between the PCR primers increases exponentially as the number of primers included in the reaction mixture increases. This interaction usually results in preferential amplification of unwanted 'primer-dimer' artifacts instead of the intended DNA templates (amplicons). Another problem in multiplex PCR is sequence-dependent differences in PCR efficiency between the amplicons. The problems of multiplexing can be reduced to some extent by using PCR primers that are as similar as possible to one another13,14,15. But the multiplexing level that can be readily achieved in standard PCRs does not reach the multiplexing capacity offered by current technology for producing high-density DNA microarrays. Simultaneous analysis of a reasonable amount of genomic DNA with the current detection sensitivity of microarray scanners requires an amplification step. The PCR step complicates the molecular reaction principles underlying the assays and introduces multiple laboratory steps into the procedures and is therefore the chief obstacle to highly multiplexed SNP genotyping.
Complexity reduction. One approach taken to reduce the complexity of the genome before SNP genotyping is cleavage of genomic DNA with a restriction enzyme, ligation of common adaptor sequences to the restriction fragments and use of the adaptor sequences as binding sites for a single universal PCR primer16. This principle, which allows the amplification of thousands of DNA fragments with one PCR primer pair in a single reaction, was pioneered more than 10 years ago for generating fingerprints of anonymous plant DNA fragments using gel electrophoresis in the amplified fragment-length polymorphism technique17. The same principle has also been used to reduce the complexity of the genome before SNP discovery by sequencing18 and by hybridization to high-density oligonucleotide arrays19. The complexity of the human genome can be reduced by a factor of 50 (to 60 Mb) by using a single restriction enzyme (XbaI) followed by PCR with a universal primer to amplify fragments 250–1,000 bp in size20. This combined complexity-reduction and target-amplification step enables the genotyping of 10,000 SNPs using allele-specific hybridization to probes immobilized on high-density oligonucleotide arrays (GeneChip 10K arrays). One drawback of this PCR strategy is that only preselected panels of SNPs located in the amplified fragments can be genotyped. The multiplexing level of this system has been further increased to 100,000 SNPs by using a combination of two enzymes (XbaI and HindIII) for the complexity-reduction step and by amplifying PCR fragments up to 2,000 bp in two separate reactions21 (GeneChip 100K arrays). Here, ∼10% (300 Mb) of the genome is represented in the amplified fragments, which would correspond to multiplexed amplification of 100,000 DNA fragments in only two PCR reactions.
Long-range PCR. The high-density–array genotyping system from Perlegen Sciences takes a 'brute force' PCR approach. This system is first described for SNP discovery and haplotyping on chromosome 21. Total genomic DNA (in haploid human-rodent cell hybrids) representing the entire chromosome 21 (1% of the genome, 32 Mb) was amplified by long-range PCR of fragments of ∼10 kb, using more than 3,000 individual PCR reactions22. The amplified fragments were resequenced by allele-specific hybridization to very large microarrays carrying oligonucleotide probes produced by photolithographic in situ synthesis23. The same approach has been used for SNP discovery across the whole genome and for the genotyping of more than 2 million SNPs in (diploid) human genomic DNA samples5. The coverage of the human genome was 92% and required long-range PCR of 300,000 fragments. The amplicons were genotyped for 2 million SNPs using 49 different high-density oligonucleotide arrays for each individual sample. Also in this case, the complexity of the genomic DNA applied to an array is then reduced by a factor of ∼50.
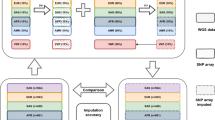
PCR for signal amplification. Carrying out the SNP-genotyping reaction directly on genomic DNA without a prior amplification step, and using a PCR amplification step only for increasing the number of detectable target molecules created by the genotyping reaction, is a reverse and more flexible strategy for addressing the problem of multiplexing the PCR. 'Circularizable' probes, originally called 'padlock' probes24, that contain two regions complementary to adjacent regions in the target DNA sequence are allowed to anneal directly to the genomic DNA, followed by enzyme-assisted detection of the SNP alleles in the Molecular Inversion Probe assay (Parallele Biosciences)25. Analogously, in the GoldenGate assay (Illumina), two oligonucleotides are used for recognition of the target DNA flanking the SNPs prior to enzyme-assisted genotyping26,27. The genotyping reactions generate templates that can be amplified by PCR using universal primer sequences for which binding sites have been inserted by means of the detector oligonucleotides in both assays (Fig. 1). Tag sequences, introduced into the templates using the probes and primers, are used to capture the allele-specific PCR products on GeneChip (Affymetrix) or BeadArrays (Illumina) that carry immobilized complementary tag sequences. Proof of principle for the Molecular Inversion Probe assay was first shown by multiplexed genotyping of 1,500 SNPs25. Further refinement of the system with improved algorithms for assay design and genotype assignment have recently allowed multiplexing levels exceeding 10,000 SNPs28. The GoldenGate assay can be multiplexed for genotyping panels of 1,500 SNPs27. Both the Molecular Inversion Probe and the GoldenGate assays permit genotyping of custom-designed SNP panels from any genomic region of interest. A recently devised modification of the principles underlying the GoldenGate assay opens up the possibility of genome-wide genotyping of SNPs directly in the complexity of the whole genome without using a PCR step. A large number (106–107) of fragmented copies of the genome are initially produced by whole-genome amplification with random primers in this assay. A signal amplification procedure is used to attain sufficient fluorescence-detection sensitivity after genotyping. Proof of principle for the whole-genome genotyping assay was demonstrated by comparing genotyping data for a panel of 1,500 SNPs with genotyping data for the same panel of SNPs obtained by the original GoldenGate assay29. This principle for whole-genome genotyping is the basis for a new system for genotyping a genome-wide panel of 100,000 'exon-centric' SNPs (Illumina).

Reaction principles and steps of the Molecular Inversion Probe assay (a) and the GoldenGate assay (b) illustrated for one heterozygous A/G SNP. (b) The genomic DNA is biotinylated and immobilized on avidin-coated microparticles. In both assays, oligonucleotides containing target-specific regions (red), binding sites for PCR primers (blue) and tag sequences for capture on a solid support (green) are allowed to anneal directly to genomic DNA. The 3′ end of the upstream target-specific oligonucleotide is extended by a DNA polymerase, followed by ligation to the downstream oligonucleotide. (a) The probe is extended by a single nucleotide in two separate single-base reactions. (b) A pair of allele-specific primers is used. The new, allele-specific DNA molecules created by the reactions are amplified by PCR. (a) The circular molecule is cleaved at a site between the PCR primers before and after PCR, followed by fluorescent labeling. (b) The fluorescence labels are introduced using the PCR primers. The labeled molecules are captured on GeneChip glass microarray (a) or a BeadArray (b) carrying complementary tag sequences for fluorescence detection.
Reaction principles and microarray design
Surprisingly, the microarray-based SNP genotyping systems used today rely on molecular strategies for distinction between SNP alleles that were introduced about 15 years ago12,30,31,32,33. The reaction principles and assays used for SNP genotyping have been reviewed elsewhere34,35. The robustness of the multiplexed microarray-based SNP genotyping systems is determined by the reaction principles applied for SNP allele distinction and the microarray formats used, given that the issues related to PCR have been addressed as discussed above. Table 1 summarizes some of the technical features of the current microarray-based SNP-genotyping systems. In addition, computer software for designing probes and primer for the assays, and algorithms for assigning the genotypes, are essential elements in each of the systems.
Allele-specific hybridization. Hybridization with allele-specific oligonucleotide probes (ASOs) is the reaction principle underlying the GeneChip assays. In allele-specific hybridization, the difference in thermal stability between a perfectly matched and mismatched ASO probe and its DNA target is used to distinguish between the SNP alleles. The thermal stability of an ASO probe hybrid depends to a large extent on the nucleotide sequence flanking a SNP, in addition to the stringency (temperature and ionic strength) of the reaction conditions. This intrinsic property of allele-specific hybridization impedes the design of multiplexed ASO microarrays for genotyping any SNP. In the GeneChip systems, this problem is circumvented by careful selection of the SNPs included in the panels on the basis of their performance in the assays and by interrogating each SNP using 40 different ASO probes. The arrays are designed to carry four sets of ten ASO probes corresponding to both strands of the SNP alleles with matched and mismatched probes for each SNP position and four additional nucleotide positions flanking the SNP5,20,21. The requirement of multiple probes per SNP results in very large microarrays for highly multiplexed genotyping. Two arrays with a probe feature size of 8 μM carrying 2.5 million different probes per array are used to genotype 100,000 SNPs in each individual DNA sample21. Correspondingly, as many as 49 such arrays are needed to genotype 2 million SNPs in one sample5. Manufacturing of arrays with oligonucleotides at such high density is accomplished by photolithographic techniques based on combinatorial synthesis of the probes in situ on the arrays23,36. The genotypes are assigned on the basis of the joint fluorescence patterns generated by the 40 hybridization reactions for each SNP using classification- or model-based algorithms developed specifically for this purpose37,38.
DNA polymerase- and ligase-assisted genotyping. Enzyme-assisted SNP genotyping methods provide highly specific distinction between SNP alleles34. This can be explained by the high accuracy of nucleotide incorporation by the DNA polymerases or the high specificity of the DNA ligases in joining two adjacent and perfectly matched DNA strands. Single-base primer extension and ligation are essentially independent of the DNA sequence context and therefore allow genotyping of most SNPs in the same reaction conditions. This possibility provides an obvious advantage compared with allele-specific hybridization by reducing the number of oligonucleotides required in the microarrays systems for multiplexed SNP genotyping. The first microarray-based systems relying on primer extension with SNP-specific primers39,40,41 or ligation probes immobilized on array surfaces42 were devised several years ago. The use of generic microarrays with complementary 'tag' or 'barcode' sequences for capturing the products of enzyme-assisted SNP genotyping reactions done in solution allows flexible design of the SNP assays and simplifies manufacturing of the microarrays. The concept of using tag sequences was first applied for PCR-based analysis of expressed yeast sequences43 using GeneChip arrays. For SNP genotyping, tag arrays have been used in single-base primer extension assays in combination with both high-density44 and lower-density45,46,47 microarrays, with microparticles48,49 and with ligase-assisted detection50,51.
In the Molecular Inversion Probe assay25, hybridization of the two ends of the probes to their genomic DNA targets leaves a single-base gap between the probe ends. The gap is filled by a single-base primer extension reaction, which can distinguish between the SNP alleles, and followed by circularization of the probe by joining its ends by ligation. Excess linear DNA is removed from the reaction mixture by exonuclease treatment. The circular probe molecule, containing primer-binding sites, serves as a template for inverse PCR, as discussed in the previous section. The amplified, inverted probe molecules are cleaved after PCR and labeled using either two or four fluorophores. They are then captured on a GeneChip tag microarray for detection using an array scanner compatible with the GeneChip format. The details of the labeling procedures used in the assays are not disclosed in the published article28. Improved design of the tag sequences was a key element for increasing the multiplexing level of the Molecular Inversion Probe assay from 1,500 SNPs to more than 10,000 SNPs28. Figure 1a illustrates the steps of the Molecular Inversion Probe assay.
In the GoldenGate assay, the required specificity for SNP genotyping is achieved by first attaching the genomic DNA to a solid support, to facilitate stringent washing procedures, and then using two oligonucleotides to recognize the genomic target regions. PCR primer-binding sites and tag sequences are also introduced using these oligonucleotides27. Allele-specific extension of the upstream primer is used to distinguish SNP alleles, followed by ligation of the extended primer to the downstream probe, creating a molecule that can be amplified by PCR. Allele-specific fluorescent labels are introduced using the PCR primers, and the reaction products are captured on BeadArrays, in which each bead type carries a unique complementary tag sequence. The BeadArray matrices have been produced by a random assembly of beads that are 0.3 μM in diameter, to which all the capturing tag sequences have been chemically coupled in a single, pooled reaction. The beads form random arrays and are decoded after production using a combinatorial hybridization scheme52. Figure 1b illustrates the steps of the GoldenGate assay. In the whole-genome genotyping assay, the randomly amplified fragments representing total genomic DNA are captured on BeadArrays carrying a pair of sequence-specific and allele-specific oligonucleotides for each SNP being genotyped. In this system, the beads carrying the decoded oligonucleotides have been grafted on Bead Chips on a microscope slide and allow genotyping of the set of 100,000 SNPs on one slide. The SNPs are genotyped by allele-specific extension of the immobilized capturing probe primers. Biotin residues are introduced during the primer-extension reaction to facilitate an indirect signal amplification step that introduces a fluorescence label for detection29.
Current and future applications of microarray-based SNP genotyping
Successful association studies for identification of alleles associated with disease susceptibility with modest effects will require the analysis of thousands of samples53. Therefore, the utility of microarray-based SNP genotyping systems in large-scale association studies is determined not only by the SNP multiplexing levels, but also by the number of samples that can be processed in parallel, and by the reagent costs involved in the assays. It has not been feasible so far, as illustrated by the comparison of SNP multiplexing levels and sample throughput between the current microarray-based SNP genotyping systems in Figure 2, to combine a high SNP multiplexing level with a high sample throughput. In the GeneChip 10K and Molecular Inversion Probe systems, for example, genotyping of 10,000 SNPs requires one microarray per DNA sample20,28. Genotyping of 2 million SNPs requires a 'wafer' of as many as 49 arrays for each individual DNA sample5. At the other extreme of the scale of SNP multiplexing and sample throughput is the SNPstream system (Beckman Coulter), which is based on tag microarrays in a glass-bottom, 384-well microtiter plate format and allows multiplex genotyping of 12 SNPs (and possibly 48 SNPs in the near future) in 384 samples on one array47. The GoldenGate assay is a functional compromise between multiplexing level and sample throughput; it allows multiplex genotyping of 1,500 SNPs in 96 samples on a single BeadArray matrix that matches a 96-well microtiter plate27.

In each of the systems, multiple arrays can be analyzed in parallel; thus, there is overlap between the methods of choice for a particular application.
The microarray-based SNP-genotyping systems have already found some medical applications despite their limitations in sample throughput and their high reagent costs per sample (Table 2). Genome-wide linkage mapping of genes using microarray systems with fixed SNP panels is a cost-effective and time-saving alternative to linkage mapping with multiallelic markers and allele separation by capillary electrophoresis. Several recent studies have found that SNP panels provide higher data quality, more accurate genotyping results and higher information content, and may also have higher power to detect linkage, than traditionally used panels of microsatellite markers54,55,56. Genome-wide detection of copy-number changes and allelic imbalance due to genomic instability in tumors is another application in which SNP genotyping systems can improve the resolution of microsatellite markers and of comparative genomic hybridization57,58. The number of samples available for analysis is typically lower for linkage studies and detection of copy-number changes in tumors than for association studies of cases and controls. So far, most association studies in large sample sets have used traditional SNP-genotyping assays in formats other than the microarray format, such as the Invader assay15,59 or single-base extension with fluorescence polarization detection60,61. One way to circumvent the limited sample throughput of the microarray systems in association studies is to pool the samples of cases and controls and detect the differences in SNP allele frequencies by quantitative genotyping in the pooled samples. This approach was recently used to screen 7,000 SNPs in candidate genes spanning 17 Mb of DNA for association with high-density lipoprotein–cholesterol levels using long-range PCR and high-density oligonucleotide arrays62.
A considerable advantage of the GoldenGate and Molecular Inversion Probe assays is the flexibility of SNP selection. The most important application of these systems so far has been the production of highly accurate genotype data for more than 800,000 SNPs in the International Haplotype Mapping Project4. Because these genotype data are placed in public databases, they can serve as a resource for the development of statistical methods for selecting the most informative tagging SNPs and for population studies on LD and haplotype structure. The formats of these assays are cost-effective compared with previously available SNP-genotyping systems for fine mapping of LD patterns63. They are also useful for fine mapping of genes associated with disease susceptibility in regions previously identified by linkage analysis in affected families, as well as for association studies of candidate genes. Several genotyping centers that have recently acquired high-throughput microarray-based systems have initiated studies to identify positional or functional candidate genes that predispose to complex diseases.
Selection of the most informative SNP panels for genome-wide association studies still awaits the possible results of the Haplotype Mapping projects. The number of SNPs and the optimal SNP selection for analysis will vary between different populations and genomic regions, but several hundreds of thousand of SNPs will probably be required3,7,53. To increase the possibilities of identifying functional genetic variants, a more feasible approach for genome-wide association studies may be to focus on SNPs in coding or conserved regions of the genome. The goal of genome-wide association studies does not seem as distant as it was when this approach was first proposed almost ten years ago2 because of the recent development of highly multiplexed SNP methods. But there are still technical issues related to sample throughput and assay cost, as well as issues related to the statistical analysis of the data, that need to be addressed before comprehensive genome-wide association studies can be implemented in practice to identify genetic variants that cause common complex diseases and traits.
References
Kruglyak, L. & Nickerson, D.A. Variation is the spice of life. Nat. Genet. 27, 234–236 (2001).
Risch, N. & Merikangas, K. The future of genetic studies of complex human diseases. Science 273, 1516–1517 (1996).
Hirschhorn, J.N. & Daly, M.J. Genome-wide association studies for common diseases and complex traits. Nat. Rev. Genet. 6, 95–108 (2005).
The International HapMap Project. Nature 426, 789–796 (2003).
Hinds, D.A. et al. Whole-genome patterns of common DNA variation in three human populations. Science 307, 1072–1079 (2005).
Johnson, G.C. et al. Haplotype tagging for the identification of common disease genes. Nat. Genet. 29, 233–237 (2001).
Evans, D.M., Cardon, L.R. & Morris, A.P. Genotype prediction using a dense map of SNPs. Genet. Epidemiol. 27, 375–384 (2004).
Crawford, D.C. et al. Haplotype diversity across 100 candidate genes for inflammation, lipid metabolism, and blood pressure regulation in two populations. Am. J. Hum. Genet. 74, 610–622 (2004).
Segal, E., Friedman, N., Kaminski, N., Regev, A. & Koller, D. From signatures to models: understanding cancer using microarrays. Nat. Genet. 37, 38–45 (2005).
Pinkel, D. & Albertson, D.G. Array comparative genomic hybridization and its applications in cancer. Nat. Genet. 37, 11–17 (2005).
Mullis, K.B. & Faloona, F.A. Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods Enzymol. 155, 335–350 (1987).
Saiki, R.K., Walsh, P.S., Levenson, C.H. & Erlich, H.A. Genetic analysis of amplified DNA with immobilized sequence-specific oligonucleotide probes. Proc. Natl. Acad. Sci. USA 86, 6230–6234 (1989).
Lin, Z., Cui, X. & Li, H. Multiplex genotype determination at a large number of gene loci. Proc. Natl. Acad. Sci. USA 93, 2582–2587 (1996).
Brownie, J. et al. The elimination of primer-dimer accumulation in PCR. Nucleic Acids Res. 25, 3235–3241 (1997).
Ohnishi, Y. et al. A high-throughput SNP typing system for genome-wide association studies. J. Hum. Genet. 46, 471–7 (2001).
Kennedy, G.C. et al. Large-scale genotyping of complex DNA. Nat. Biotechnol. 21, 1233–1237 (2003).
Vos, P. et al. AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res. 23, 4407–4414 (1995).
Altshuler, D. et al. An SNP map of the human genome generated by reduced representation shotgun sequencing. Nature 407, 513–516 (2000).
Dong, S. et al. Flexible use of high-density oligonucleotide arrays for single-nucleotide polymorphism discovery and validation. Genome Res. 11, 1418–1424 (2001).
Matsuzaki, H. et al. Parallel genotyping of over 10,000 SNPs using a one-primer assay on a high-density oligonucleotide array. Genome Res. 14, 414–425 (2004).
Matsuzaki, H. et al. Genotyping over 100,000 SNPs on a pair of oligonucleotide arrays. Nat. Methods 1, 109–111 (2004).
Patil, N. et al. Blocks of limited haplotype diversity revealed by high-resolution scanning of human chromosome 21. Science 294, 1719–1723 (2001).
Fodor, S.P. et al. Light-directed, spatially addressable parallel chemical synthesis. Science 251, 767–773 (1991).
Nilsson, M. et al. Padlock probes: circularizing oligonucleotides for localized DNA detection. Science 265, 2085–2088 (1994).
Hardenbol, P. et al. Multiplexed genotyping with sequence-tagged molecular inversion probes. Nat. Biotechnol. 21, 673–678 (2003).
Oliphant, A., Barker, D.L., Stuelpnagel, J.R. & Chee, M.S. BeadArray technology: enabling an accurate, cost-effective approach to high-throughput genotyping. Biotechniques Suppl., 56–58, 60–61 (2002).
Fan, J.B. et al. Highly parallel SNP genotyping. Cold Spring Harb. Symp. on Quant. Biol. LXVII, 69–78 (2003).
Hardenbol, P. et al. Highly multiplexed molecular inversion probe genotyping: Over 10,000 targeted SNPs genotyped in a single tube assay. Genome Res. 15, 269–275 (2005).
Gunderson, K.L., Steemers, F.J., Lee, G., Mendoza, L.G. & Chee, M.S. A genome-wide scalable SNP genotyping assay using microarray technology. Nat. Genet. 37, 549–554 (2005).
Landegren, U., Kaiser, R., Sanders, J. & Hood, L. A ligase-mediated gene detection technique. Science 241, 1077–1080 (1988).
Newton, C.R. et al. Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS). Nucleic Acids Res. 17, 2503–2516 (1989).
Syvänen, A.-C., Aalto-Setala, K., Kontula, K. & Söderlund, H. A primer-guided nucleotide incorporation assay in the genotyping of apolipoprotein E. Genomics 8, 684–692 (1990).
Southern, E.M., Maskos, U. & Elder, J.K. Analyzing and comparing nucleic acid sequences by hybridization to arrays of oligonucleotides: evaluation using experimental models. Genomics 13, 1008–1017 (1992).
Syvanen, A.C. Accessing genetic variation: genotyping single nucleotide polymorphisms. Nat. Rev. Genet. 2, 930–942 (2001).
Kwok, P.Y. Methods for genotyping single nucleotide polymorphisms. Annu. Rev. Genomics Hum. Genet. 2, 235–258 (2001).
Pease, A.C. et al. Light-generated oligonucleotide arrays for rapid DNA sequence analysis. Proc. Natl. Acad. Sci. USA 91, 5022–5026 (1994).
Cutler, D.J. et al. High-throughput variation detection and genotyping using microarrays. Genome Res. 11, 1913–1925 (2001).
Liu, W.M. et al. Algorithms for large-scale genotyping microarrays. Bioinformatics 19, 2397–2403 (2003).
Shumaker, J.M., Metspalu, A. & Caskey, C.T. Mutation detection by solid phase primer extension. Hum. Mutat. 7, 346–354 (1996).
Pastinen, T., Kurg, A., Metspalu, A., Peltonen, L. & Syvanen, A.C. Minisequencing: a specific tool for DNA analysis and diagnostics on oligonucleotide arrays. Genome Res. 7, 606–614 (1997).
Pastinen, T. et al. A system for specific, high-throughput genotyping by allele-specific primer extension on microarrays. Genome Res. 10, 1031–1042 (2000).
Gerry, N.P. et al. Universal DNA microarray method for multiplex detection of low abundance point mutations. J. Mol. Biol. 292, 251–262 (1999).
Shoemaker, D.D., Lashkari, D.A., Morris, D., Mittmann, M. & Davis, R.W. Quantitative phenotypic analysis of yeast deletion mutants using a highly parallel molecular bar-coding strategy. Nat. Genet. 14, 450–456 (1996).
Fan, J.B. et al. Parallel genotyping of human SNPs using generic high-density oligonucleotide tag arrays. Genome Res. 10, 853–860 (2000).
Hirschhorn, J.N. et al. SBE-TAGS: an array-based method for efficient single-nucleotide polymorphism genotyping. Proc. Natl. Acad. Sci. USA 97, 12164–12169 (2000).
Lindroos, K., Sigurdsson, S., Johansson, K., Ronnblom, L. & Syvanen, A.C. Multiplex SNP genotyping in pooled DNA samples by a four-colour microarray system. Nucleic Acids Res. 30, e70 (2002).
Bell, P.A. et al. SNPstream UHT: ultra-high throughput SNP genotyping for pharmacogenomics and drug discovery. Biotechniques Suppl., 70–72, 74, 76–77 (2002).
Chen, J. et al. A microsphere-based assay for multiplexed single nucleotide polymorphism analysis using single base chain extension. Genome Res. 10, 549–557 (2000).
Cai, H. et al. Flow cytometry-based minisequencing: a new platform for high-throughput single-nucleotide polymorphism scoring. Genomics 66, 135–143 (2000).
Favis, R. et al. Universal DNA array detection of small insertions and deletions in BRCA1 and BRCA2. Nat. Biotechnol. 18, 561–564 (2000).
Baner, J. et al. Parallel gene analysis with allele-specific padlock probes and tag microarrays. Nucleic Acids Res. 31, e103 (2003).
Gunderson, K.L. et al. Decoding randomly ordered DNA arrays. Genome Res. 14, 870–877 (2004).
Wang, W.Y.S., Barratt, B.J., Clayton, D.G. & Todd, J.A. Genome-wide association studies: theoretical and practical concerns. Nat. Genet. 6, 109–118 (2005).
Murray, S.S. et al. A highly informative SNP linkage panel for human genetic studies. Nat. Methods 1, 1–5 (2004).
Evans, D.M. & Cardon, L.R. Guidelines for genotyping in genomewide linkage studies: single-nucleotide-polymorphism maps versus microsatellite maps. Am. J. Hum. Genet. 75, 687–692 (2004).
Middleton, F.A. et al. Genomewide linkage analysis of bipolar disorder by use of a high-density single-nucleotide-polymorphism (SNP) genotyping assay: a comparison with microsatellite marker assays and finding of significant linkage to chromosome 6q22. Am. J. Hum. Genet. 74, 886–897 (2004).
Koed, K. et al. High-density single nucleotide polymorphism array defines novel stage and location-dependent allelic imbalances in human bladder tumors. Cancer Res. 65, 34–45 (2005).
Zhao, X. et al. An integrated view of copy number and allelic alterations in the cancer genome using single nucleotide polymorphism arrays. Cancer Res. 64, 3060–3071 (2004).
Ozaki, K. et al. Functional SNPs in the lymphotoxin-alpha gene that are associated with susceptibility to myocardial infarction. Nat. Genet. 32, 650–654 (2002).
Chen, X. & Kwok, P.Y. Template-directed dye-terminator incorporation (TDI) assay: a homogeneous DNA diagnostic method based on fluorescence resonance energy transfer. Nucleic Acids Res. 25, 347–353 (1997).
Helgadottir, A. et al. The gene encoding 5-lipoxygenase activating protein confers risk of myocardial infarction and stroke. Nat. Genet. 36, 233–239 (2004).
Hinds, D.A. et al. Application of pooled genotyping to scan candidate regions for association with HDL cholesterol levels. Hum. Genomics 1, 421–434 (2004).
Ke, X. et al. The impact of SNP density on fine-scale patterns of linkage disequilibrium. Hum. Mol. Genet. 13, 577–588 (2004).
Kurg, A. et al. Arrayed primer extension: solid-phase four-color DNA resequencing and mutation detection technology. Genet. Test. 4, 1–7 (2000).
Schaid, D.J. et al. Comparison of microsatellites versus single-nucleotide polymorphisms in a genome linkage screen for prostate cancer-susceptibility Loci. Am. J. Hum. Genet. 75, 948–965 (2004).
John, S. et al. Whole-genome scan, in a complex disease, using 11,245 single-nucleotide polymorphisms: comparison with microsatellites. Am. J. Hum. Genet. 75, 54–64 (2004).
Saba, T.G. et al. An atypical form of erythrokeratodermia variabilis maps to chromosome 7q22. Hum. Genet. 116, 167–171 (2005).
Sigurdsson, S. et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am. J. Hum. Genet. 76, 528–537 (2005).
Acknowledgements
I thank L. Andersson, M. Jonsson and P. Lundmark for help with the references, tables and Figure 2; and L. Kurland and L. Lovmar for comments on the manuscript. The development and application of technology for SNP genotyping in my laboratory is supported by the Swedish Research Council and the Knut and Alice Wallenberg Foundation, through the Wallenberg Consortium North.
Author information
Authors and Affiliations
Ethics declarations
Competing interests
The author declares no competing financial interests.
Rights and permissions
About this article
Cite this article
Syvänen, AC. Toward genome-wide SNP genotyping. Nat Genet 37 (Suppl 6), S5–S10 (2005). https://doi.org/10.1038/ng1558
Issue Date:
DOI: https://doi.org/10.1038/ng1558
This article is cited by
-
Single nucleotide polymorphism SNP19140160 A > C is a potential breeding locus for fast-growth largemouth bass (Micropterus salmoides)
BMC Genomics (2024)
-
A duplex tetra-primer ARMS-PCR assay to discriminate three species of the Schistosoma haematobium group: Schistosoma curassoni, S. bovis, S. haematobium and their hybrids
Parasites & Vectors (2023)
-
Single nucleotide polymorphism discovery and functional annotation analysis of blackberry fruit
Trees (2022)
-
Genome-Wide Association Mapping of Dark Green Color Index using a Diverse Panel of Soybean Accessions
Scientific Reports (2020)
-
Reinventing quantitative genetics for plant breeding: something old, something new, something borrowed, something BLUE
Heredity (2020)
